1.背景
紫杉醇($Taxol$)是从红豆杉属植物中提取得到的天然抗肿瘤药物,其作用机制为促进微管聚合以抑制肿瘤细胞的有丝分裂。它是医药市场上最优秀的天然抗肿瘤药物之一,其国际医药市场需求巨大。$^{[1]}$
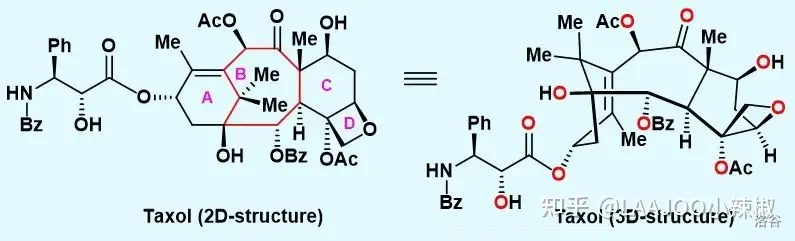
上图是紫杉醇的结构。可以看到,紫杉醇具有高度氧化的 $[6-8-6-4]$ 核心骨架,同时具有 $11$ 个手性中心(其中 $3$ 个为季碳中心)和 $1$ 个桥头双键,其高张力的双环 $[5.3.1]$ 十一烯骨架以及高度的氧化态预示着其合成具有极大的挑战性。$^{[1]}$
但是,这难不倒人类。
有机合成化学家们从上个世纪八十年代开始探索紫杉醇的全合成。从 $1980$ 年至今,已有 $40$ 余年,超过 $60$ 个课题组参与其中。在此期间,一共开发了十几条不同的全合成路线。$1994$ 年,美国 $Scripps$ 研究所的 $Nicolaou$ 课题组在世界上首次报道了紫杉醇的全合成。$^{[1]}$
近日,南科大李闯创教授团队在 $JACS$ 上报道了抗癌明星分子紫杉醇的全合成,整条路线仅需要分离 $19$ 个中间体($21$ 步),高效简洁地完成了顶级难度的复杂天然产物紫杉醇的不对称全合成。$^{[2]}$李闯创团队作为国内首家完成紫杉醇全合成的团队,提供的合成路线也是迄今为止($2023$ 年末)最短的紫杉醇全合成路线,总体效率相比之前的方案有大幅度提高。$^{[1]}$
接下来,我们将会详细分析这一条合成路线。
观前提醒:本文中关于紫杉醇全合成路线的详解为个人研究结果,若有错误请指出。不胜感谢!
2.概要
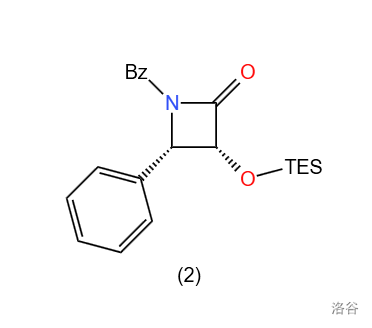
其中 $(2)$ 代表的物质为:
(图片较小可能看不清,建议$Ctrl+$滚轮放大后查看,或在新窗口打开这张图片)
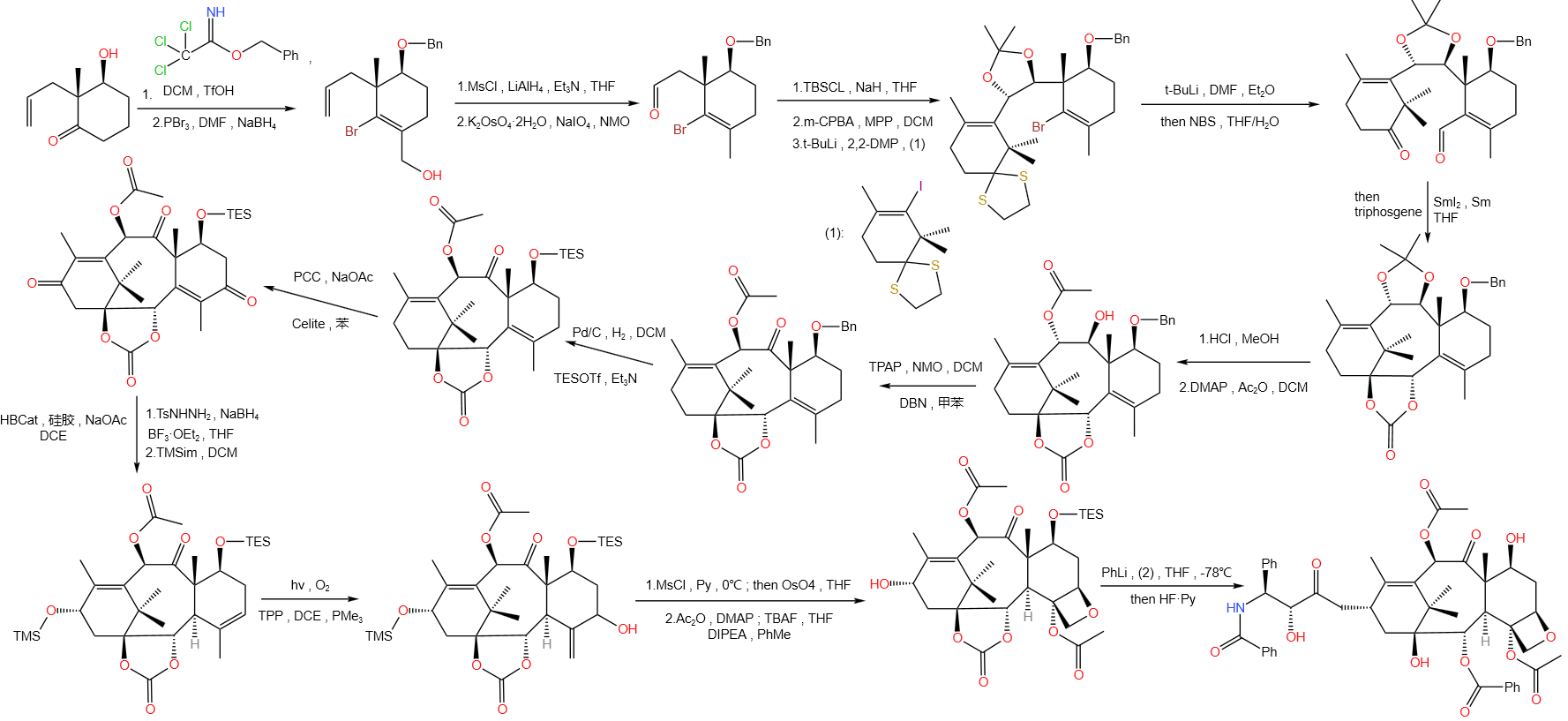
紫杉醇的全合成的难点和重点在于采用何种策略来构建八元环。李闯创教授团队在借鉴前人的合成经验的基础上,首次通过构建 $C1-C2$ 键来完成八元环的合成。作者以商业易得的手性化合物作为起始原料,首先经过 $6$ 步转化得到不稳定的醛化合物,然后与已知化合物发生加成反应,并再经一步转化为不饱和醛酮化合物。该化合物通过二碘化钐介导的分子内 $pinacol$(频哪醇)偶联反应立体选择性地构建了八元环,得到 $6/8/6$ 三环体系化合物,完成了紫杉醇的核心骨架合成。$^{[2]}$其次,利用仿生途径的单线态氧烯反应构建 $C5$ 位手性醇;此外通过串联反应同时实现 $C2$ 位苯甲酸酯和 $C13$ 位侧链安装,显著提升了合成效率。$^{[1]}$
值得一提的是,世界著名的天才合成化学家 $Baran$ 团队 $2020$ 年报道的紫杉醇全合成需要 $24$ 步,总合成效率是 0.001%;如今,李闯创团队的全合成需要21步,合成效率达到 0.118%。两个独立团队令人震撼的经典之作分别把紫杉醇的合成推向了新的高度。$^{[1]}$
接下来,我们将逐步拆分讨论李闯创课题组紫杉醇全合成。
3.逐步分析
上面的流程图中把 $21$ 步分成了 $13$ 大步,我们将跟着这幅图进行分析。
3.1 第一步
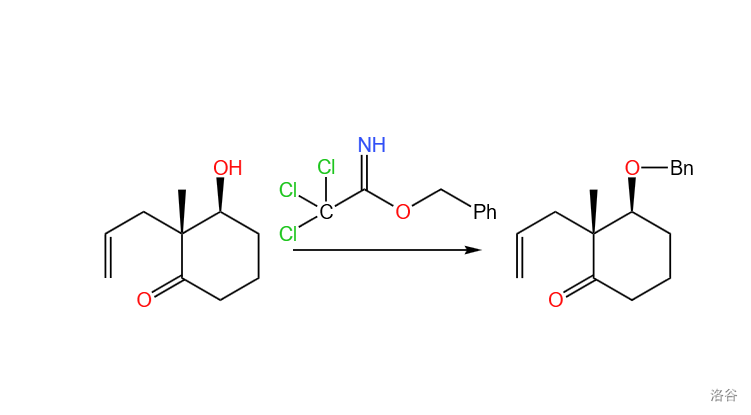
首先,通过试剂 2,2-三氯乙酰亚胺苄酯 将原料的羟基变成苄基保护起来:
然后,使用甲酰化试剂 $DMF$,溴代试剂 $PBr_3$ ,还原剂 $NaBH_4$ 一锅煮,完成第一步反应:
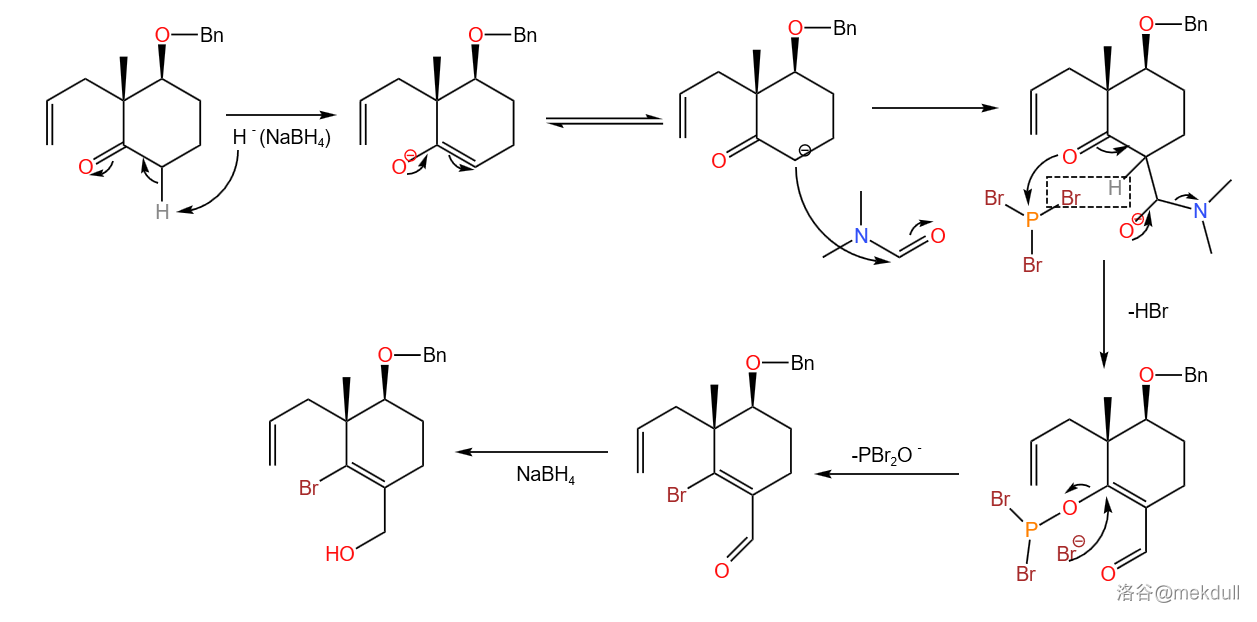
两步反应产率 $56\%$。
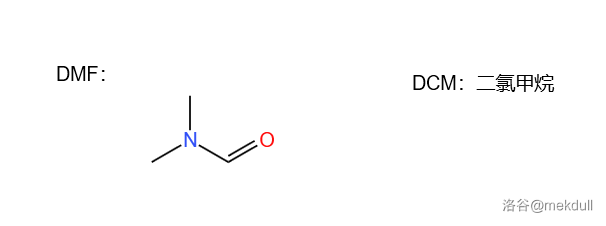
下面是本反应中用到的用英文缩写表示的试剂结构:
3.2 第二步
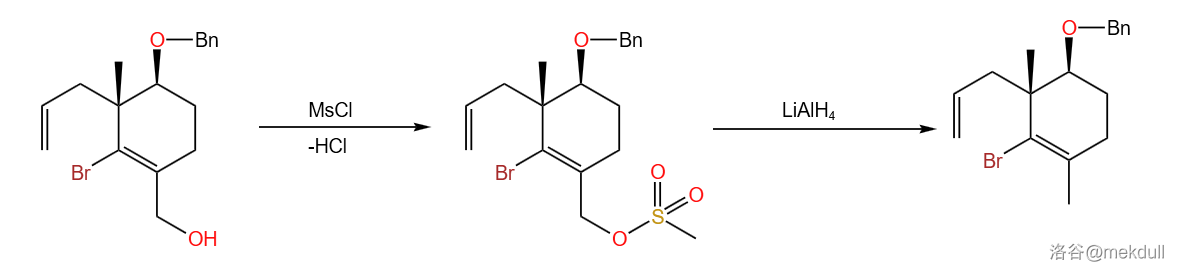
首先通过 $MsCl$ 将醇变成磺酸酯,然后用强还原剂 $LiAlH_4$ 使其发生裂解反应$^{[3]}$。由于乙烯基卤原子不活泼,所以可使其不被还原。
接着,是 Lemieux-Johnson试剂 将双键氧化开裂,即 Lemieux-Johnson反应。这是一种温和的烯烃氧化开裂法$^{[4]}$:
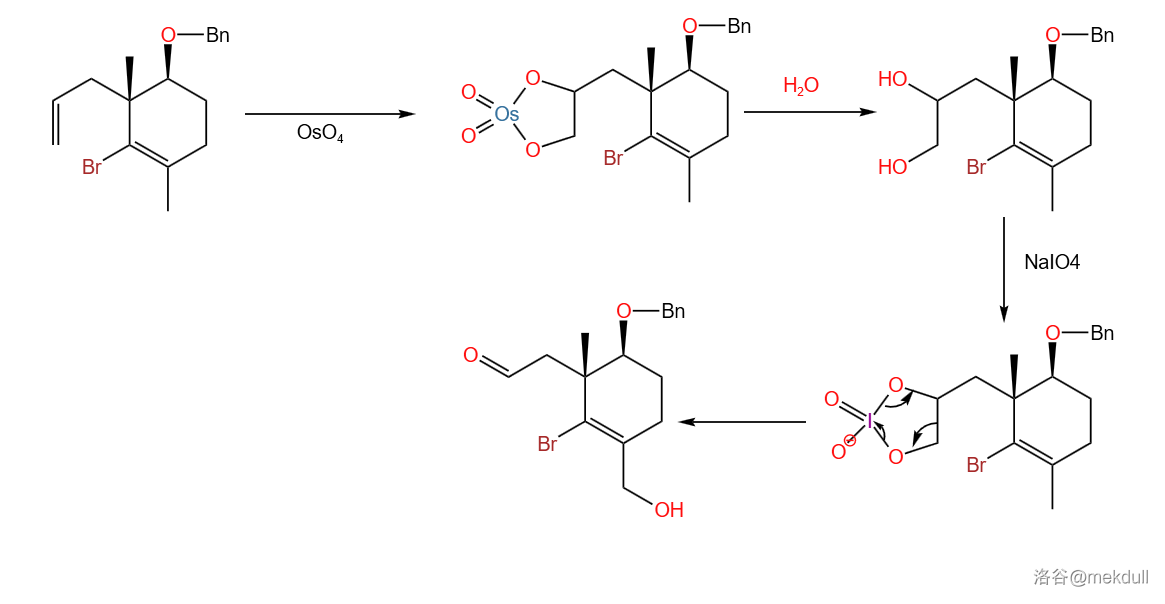
两步产率 $75\%$
本反应中用到的用英文缩写表示的试剂结构如下:
3.3 第三步
这步还是比较复杂的。其中第一小步是使用 $TBSCL$ 把醛变为烯醇硅醚$^{[5]}$,为下面的反应做准备:
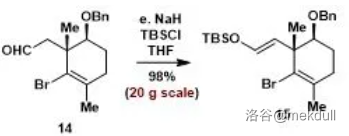
有一说一,现在的有机合成中烯醇硅醚得到了非常广泛的应用,所以确实应该学一学。这一小步反应的产率达到了 $98\%$。
第二小步是经典的 $Rubottom$ 氧化反应。使用过氧酸 $m-CPBA$ 与 $MPP$ 将烯醇硅醚 $\alpha-$ 羟基化(虽然实际得到的是被硅类保护基保护的羟基)。$Rubottom$ 氧化反应的机理首先经过一个 $Prilezhaev$ 环氧化,然后再酸性条件下开环:
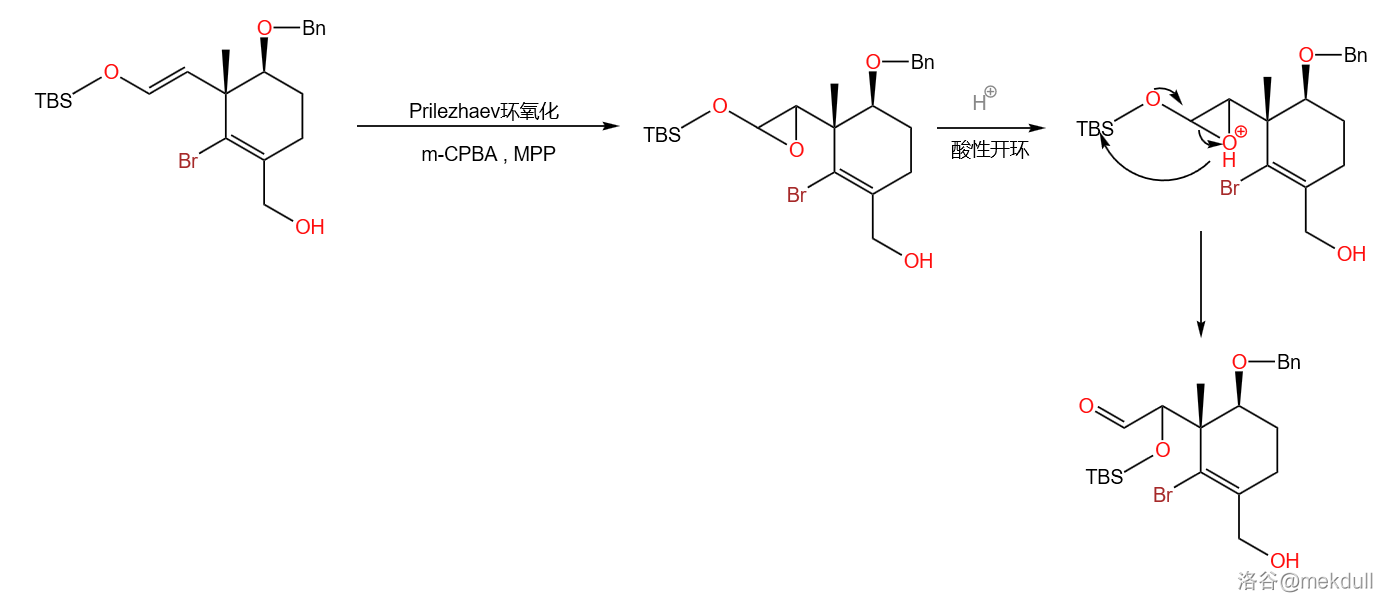
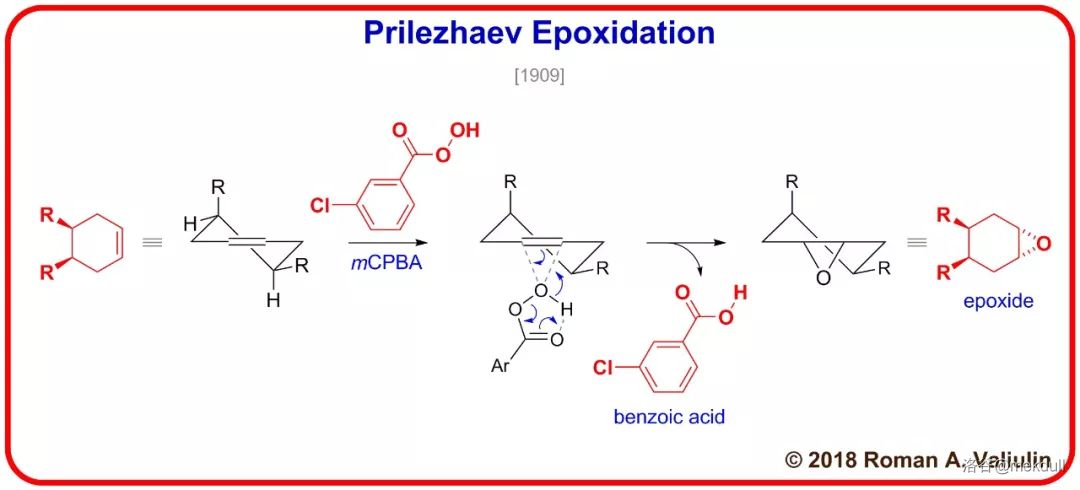
$Prilezhaev$ 环氧化的机理如下,经过蝶形过渡态:$^{[6]}$
这一小步反应产率为 $54\%$
第三小步则是关键。使用强碱将碘原子拔去生成碳负离子,再进攻羰基碳得到$1,2$ 加成产物。反应过程中还加入了 $2,2-DMP$ 对邻二醇产物进行保护:

本步中溴原子没有被拔去,可能有以下原因:
-
$C-Br$ 键比 $C-I$ 键更加牢固,难以断裂;
-
溴原子本身电负性大,难以被亲电的 $Bu^{-}$ 拔去并留下碳负离子;
-
就算拔去了,那么那个位置上生成的碳负离子也由于空间受阻很难进攻羰基;且产物为四元环,不稳定。
第三小步产率为 $76\%$
本反应中使用的由缩写表示的试剂结构如下:
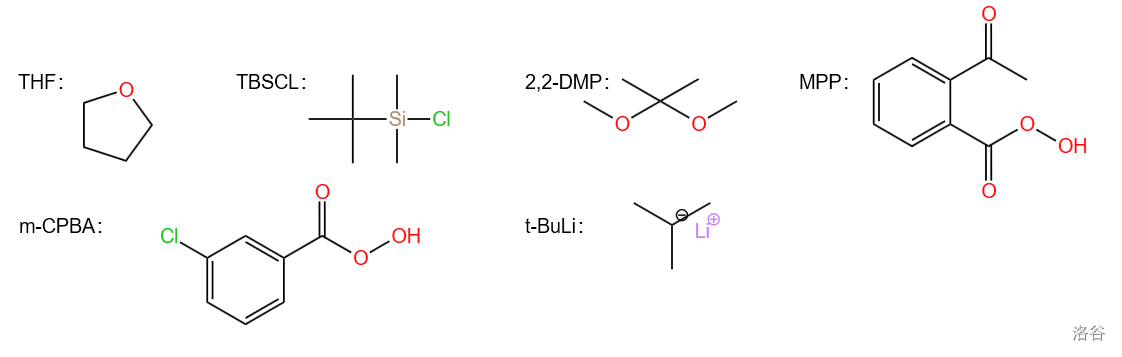
3.4 第四步
这步是将反应物再次酰基化,使用的试剂同样是 $DMF$;随后将被缩硫保护的羰基用 $NBS$ 氧化脱保护$^{[7]}$。这样便可得到两个羰基,为下一步的分子内双酮偶联($pinacol$ 偶联)做准备:
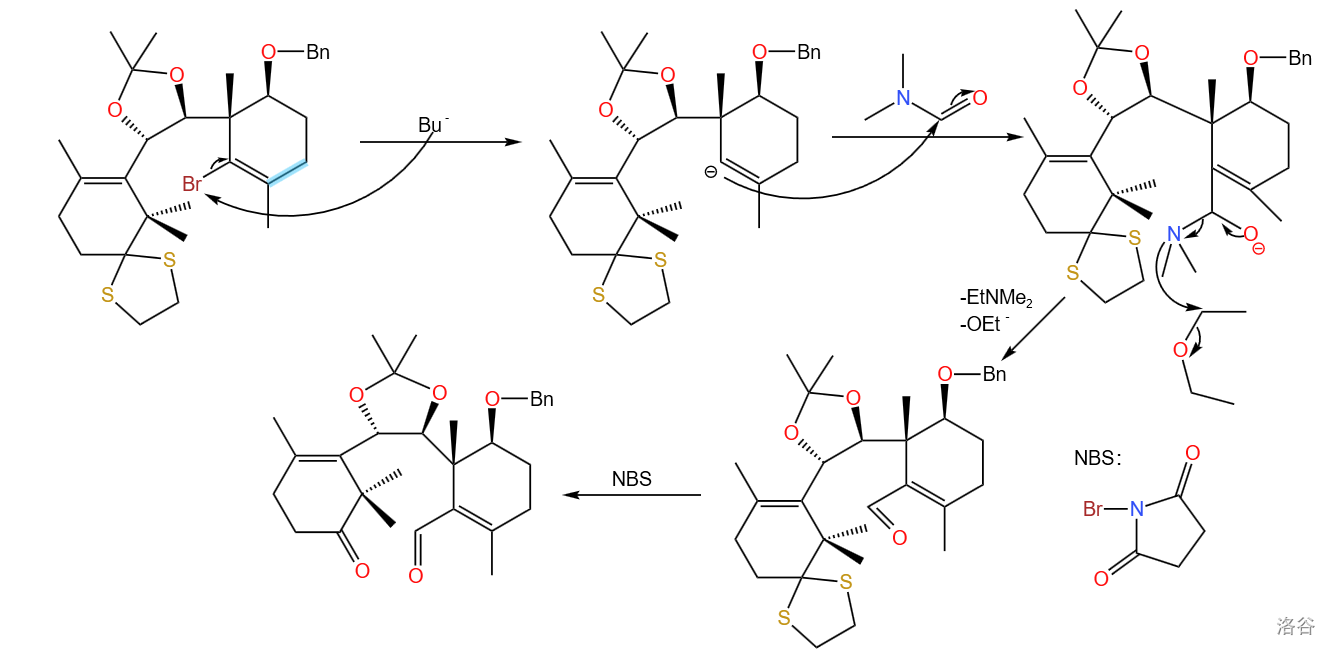
这一步产率为 $61\%$,可能是因为 $NBS$ 除了对硫代缩酮进行氧化外也可以对双键加成、对烯丙位溴代,导致了较多副产物产生。
3.5 第五步
这一步是关键中的关键。在这步中,我们将会完成紫杉醇碳骨架的构建。
在这步中,课题组采用了前所未有的策略:使用钐/二碘化钐介入的有立体选择性的 $pinacol$ 偶联$^{[9]}$在分子的 $1,2$ 位关闭八元环。随后,使用 $triphosgene$(三光气,可简写为 $BTC$)$^{[8]}$ 将生成的邻二醇变成碳酸二烷基酯保护起来:
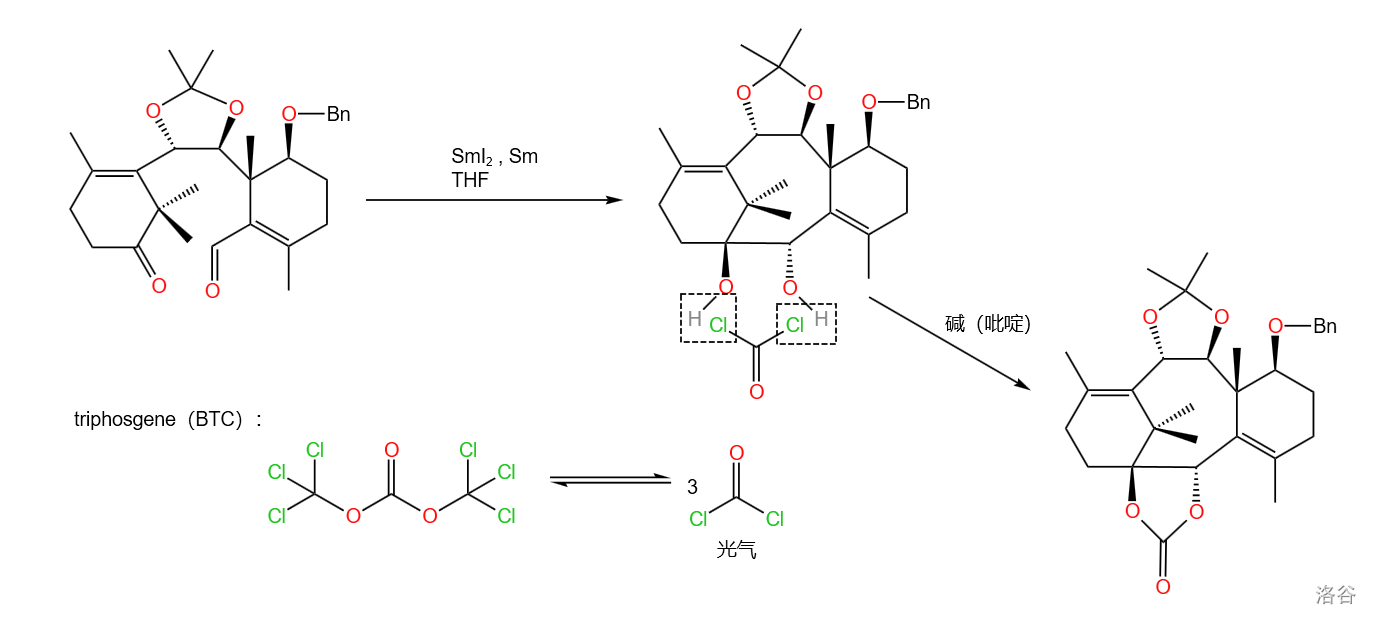
钐/二碘化钐介入的立体选择性 $pinacol$ 偶联是新近发展起来的 $C-C$ 键构筑方法,但是已经在有机物的合成中取得了很广泛的应用。
两步反应产率 $62\%$。
3.6 第六步
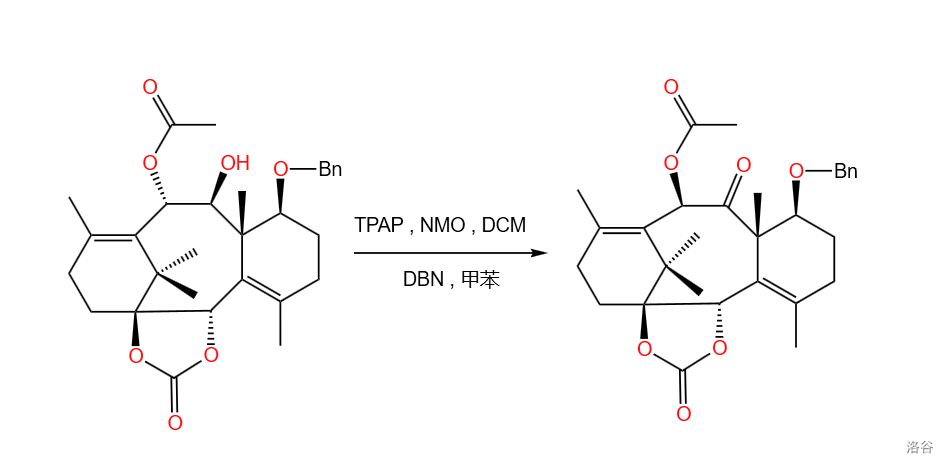
在酸性条件下,缩酮保护被脱去,随后用乙酸酐进行 $10$ 号位上的乙酰化。
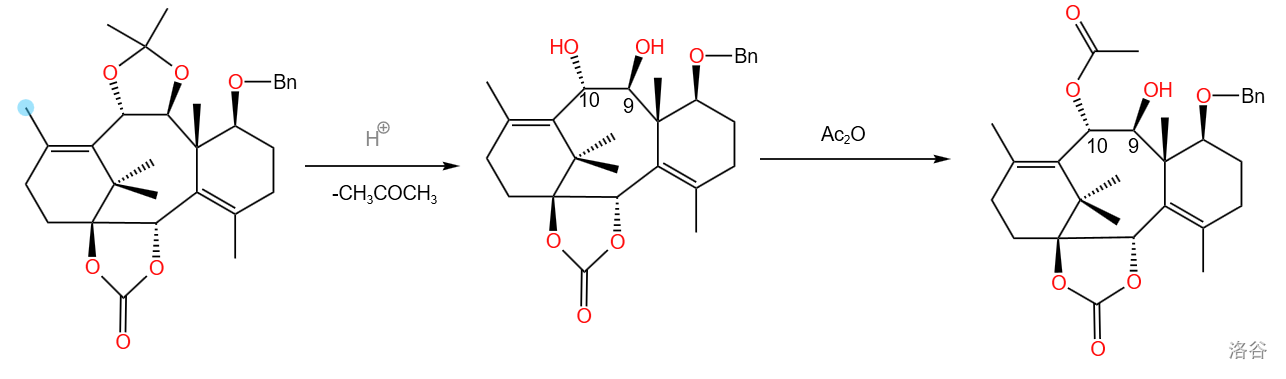
本步反应具有很高的选择性,从图中可以见到,主产物中 $9$ 号位上的羟基没有被乙酰化。这主要是反应中用到的碱性试剂 $DMAP$ 的功劳。
$DMAP$ 的结构如下:
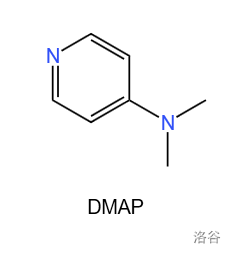
它是一种超强亲核的酰化作用催化剂,用途极为广泛。其结构上供电子的二甲氨基与母环(吡啶环)的共振,能强烈激活环上的氮原子进行亲核取代,显著地提高位阻效应$^{[10],[11]}$。在本反应中,$DMAP$ 的一个重要作用就是使得高位阻的 $9$ 号位上的羟基不被乙酰化。
该反应的产率为 $91\%$。
3.7 第七步
这一步是使用 $TPAP$(四丙基高钌酸铵)$^{[12],[13]}$对其进行氧化,将 $9$ 号位上的羟基变成羰基。
$TPAP$ 的结构如下:$^{[12]}$
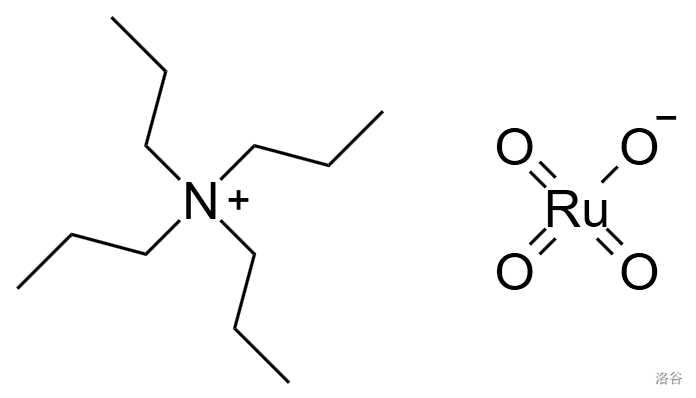
使用 $TPAP$ 的氧化反应称作 $Ley-Griffith$ 氧化。本反应中使用了 $NMO$ 协助氧化,则只需要催化量的 $TPAP$ 就可以达到同样的效果。
该反应的原理如下:$^{[13]}$
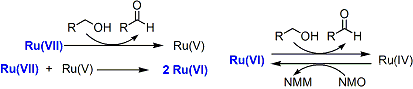
该反应的产率为 $72\%$。
3.8 第八步
这一步将 $7$ 号位上的苄基保护基换成硅类保护基 $TES$。反应可以看成先将原本的苄基保护基通过催化加氢的方法脱去$^{[14]}$,再用 $TESOTf$ 对其三乙基甲基硅烷化。$^{[15]}$
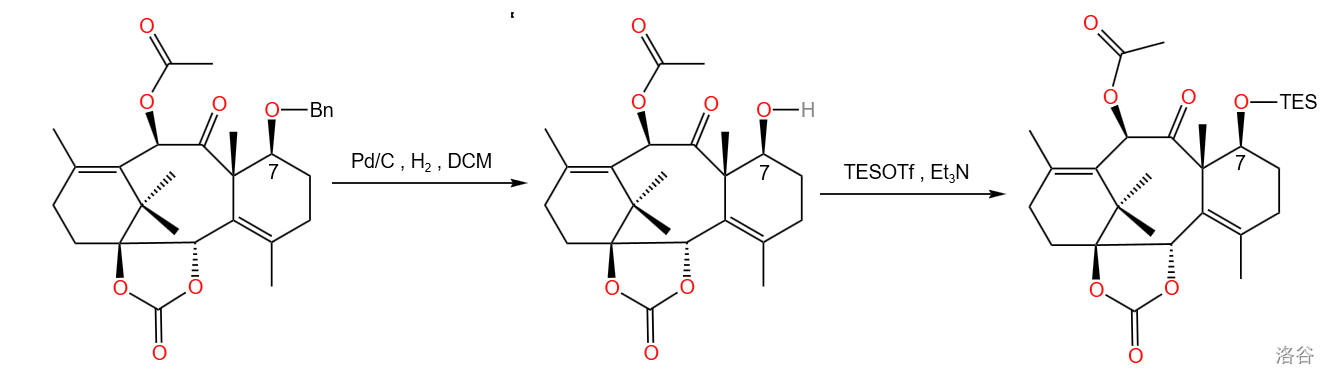
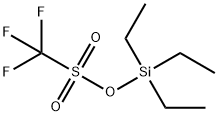
$TESOTf$ 的结构$^{[15]}$和硅基化反应的通用机理$^{[16]}$如下:
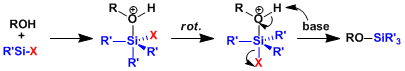
该反应的产率为 $92\%$。
3.9 第九步
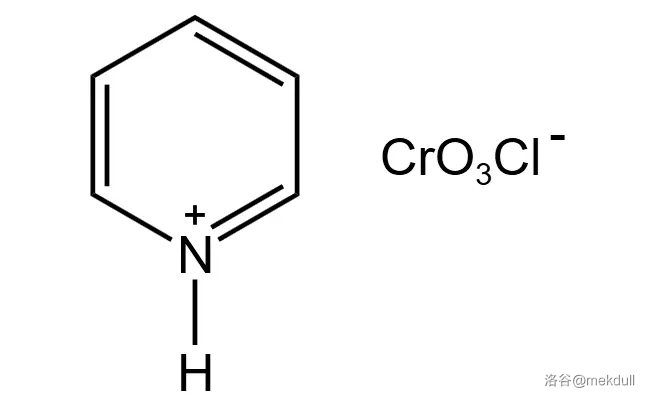
这一步是用 $PCC$ 对底物 $5$ 号位与 $13$ 号位的活泼 $C-H$ 键进行氧化$^{[17]}$(这两个位置是双键的 $\alpha$ 位,且连有两个氢原子,可以被氧化为羰基):

$PCC$ (氯铬酸吡啶)的结构如下$^{[17]}$:
该反应产率为 $75\%$
3.10 第十步
这一步比较复杂,可以分为三个小步理解。
第一小步是在 $5$ 号位上由酮制得 对甲苯磺酰腙 ,为第三小步的 二氮烯重排(即一种变异的 $Caglioti$ 反应)$^{[19,23]}$ 做准备,同时使用还原剂还原 $13$ 位上的羰基位羟基 。使用的试剂为 $NH_2NHTs$ 与 $NaBH_4$:
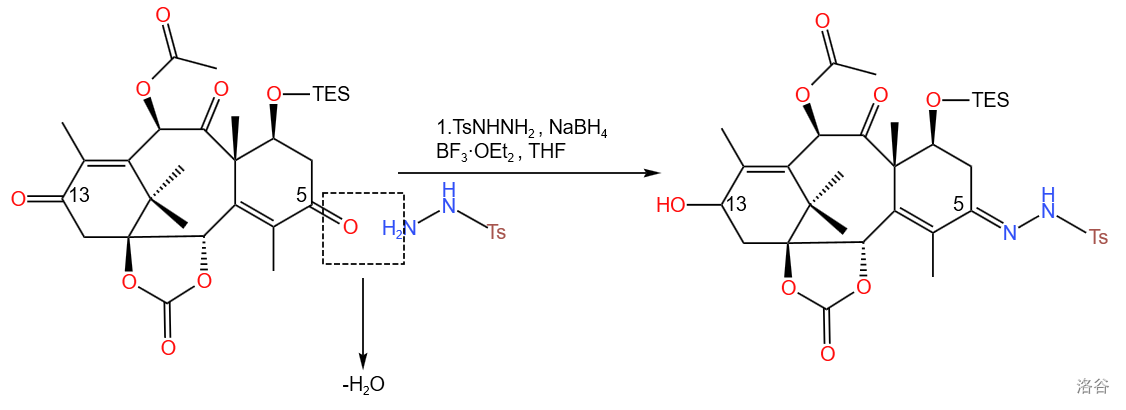
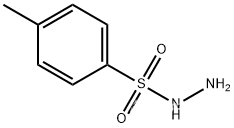
$NH_2NHTs$ 的结构如下:$^{[18]}$
该小步反应产率为 $81\%$
(其实在这里有一个我还没有解决的问题,即为什么这个反应选择性那么高。可以看到底物有很多个羰基,为什么是在 $5$ 号位上生成腙,并将 $13$ 位上的羰基还原?如果你知道为什么,请务必在评论里告诉我。)
第二小步是使用 $TMSim$ $^{[20,21]}$ 把第一小步还原出来的 $13$ 号位上的羟基变成硅醚保护起来,这也是常用的羟基保护方法:
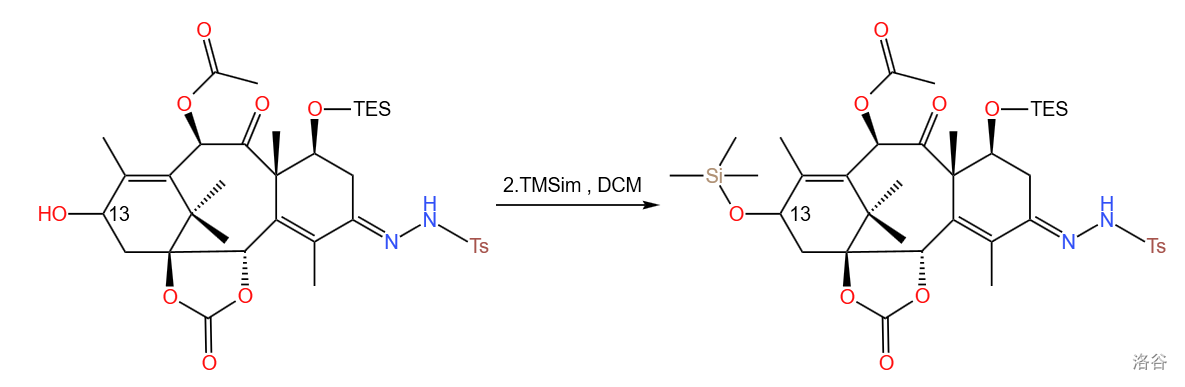
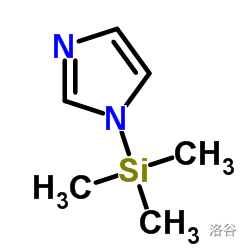
$TMSim$(三甲基硅咪唑)是硅烷化羟基的最强的硅烷化试剂,能够快速、平顺地与羟基发生反应$^{[21]}$。$TMSim$ 的结构如下:
该小步反应产率 $85\%$。
第三小步则是反应的关键。这里涉及到一个比较少见的反应:烯丙基二氮烯重排$^{[19]}$,也就是 $Caglioti$ 反应 $Kabalka$ 改进法$^{[28]}$。这是一种由 $\alpha,\beta$ 不饱和腙在 硼烷 类化合物作用下制得对应烯烃的反应。利用这个反应在 $4,5$ 位形成了双键,为下面的单线态氧烯反应做准备:
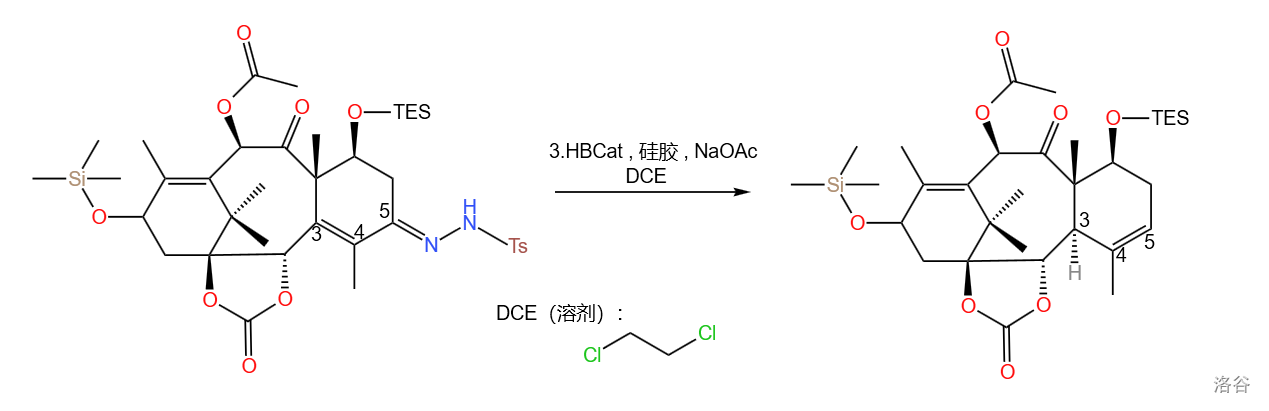
这类反应的机理如下。对于 $\alpha,\beta$ 不饱和羰基化合物来说,第二步的硼氢化是 $1,4$ 加成,所以会得到双键位移的产物:$^{[28]}$
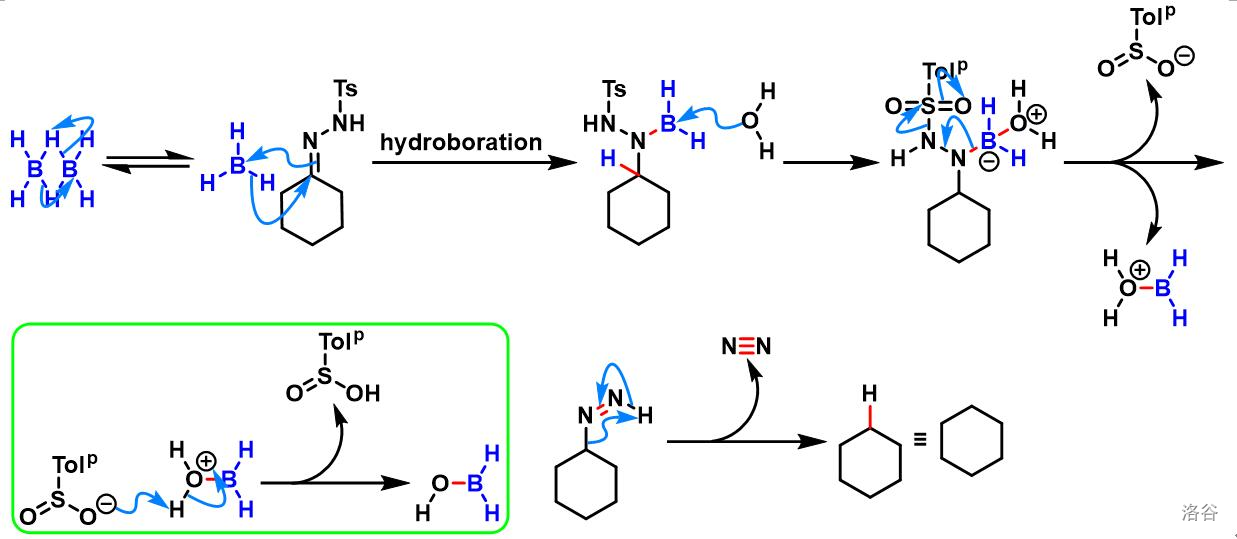
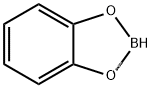
$HBCat$ 是儿萘酚硼烷的缩写$^{[22]}$,是有机反应中一个常用的还原剂。其结构如下:
该小步反应产率为 $54\%$。三小步反应总产率约为 $37\%$
3.11 第十一步
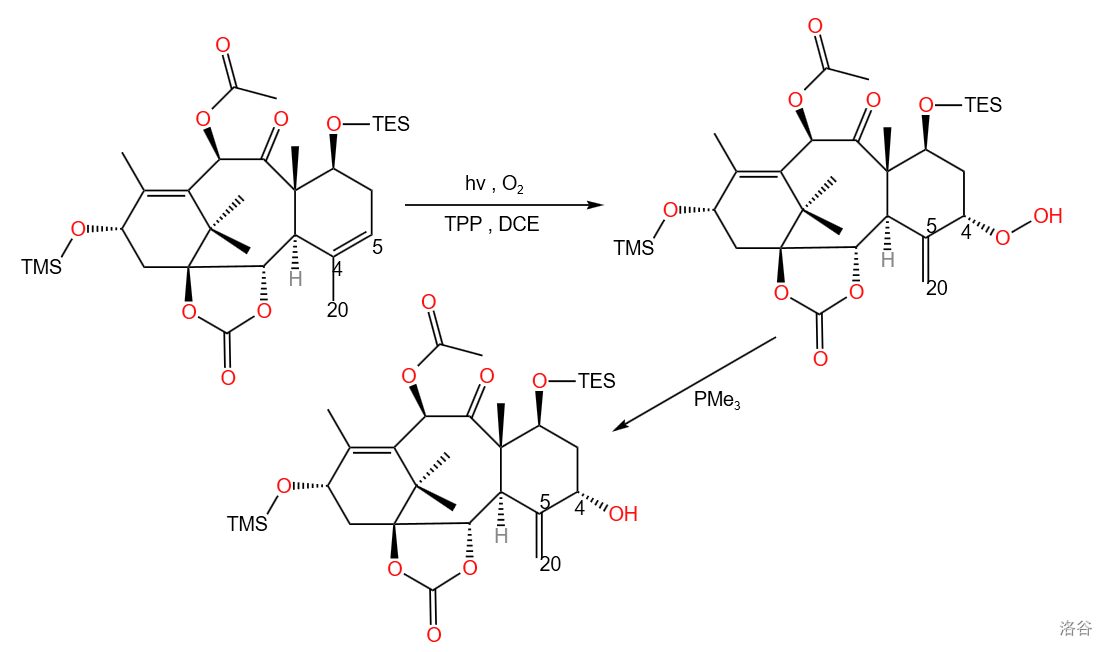
这一步反应集中体现了整条全合成路线的巧妙。使用仿生途径的 单线态氧烯反应,$^{[27]}$构筑了 $5$ 号位上的手性醇,同时将双键转移到 $4,20$ 位,为之后的烯烃顺式双羟化以及氧杂环丁烷体系构建做准备。
在光的作用下,基态氧气(三线态氧,符号为 $^{3}O_{2}$ ,也可以表示为 $^{3}\sum_{g}^{-}$)的电子被激发,得到 单线态氧($^{1}O_{2}$)。单线态氧可分为两种,即 第一单线态氧 与 第二单线态氧,符号分别为 $^{1}\Delta_{g}$ 和 $^{1}\sum_{g}^{+}$ 。它们的电子排布如下:$^{[23]}$
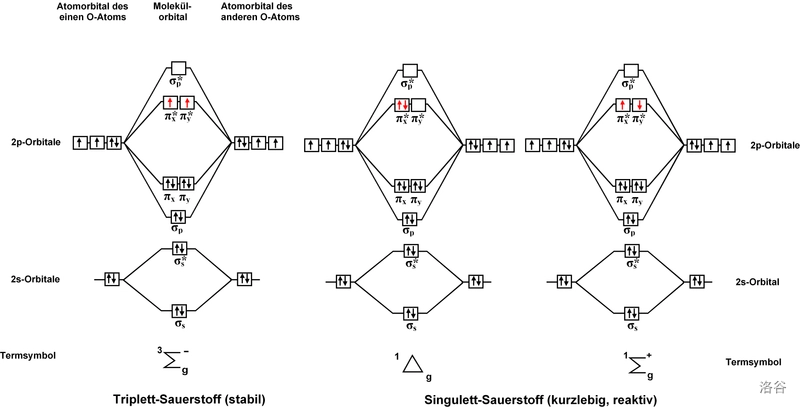
原本两个 $2pπ\ast$ 轨道中两个自旋平行的电子同时占据了一个 $2pπ\ast$ 轨道,自旋相反,形成第一单线态氧;若是占据两个 $2pπ\ast$ 轨道,自旋相反,则形成第二单线态氧。
两种单线态氧能量都高于基态,具体如下:$^{[23]}$
| 名称 | 高出基态的能量(单位:千焦每摩尔) |
|---|---|
| 第一单线态氧 | 92.0 |
| 第二单线态氧 | 154.8 |
既然单线态氧能量更高,则说明它更活泼,更容易发生反应。相比于其他 $ROS$(活性氧)$^{[24]}$,单线态氧与有机物反应时,表现出更好的选择性和更高的反应活性。
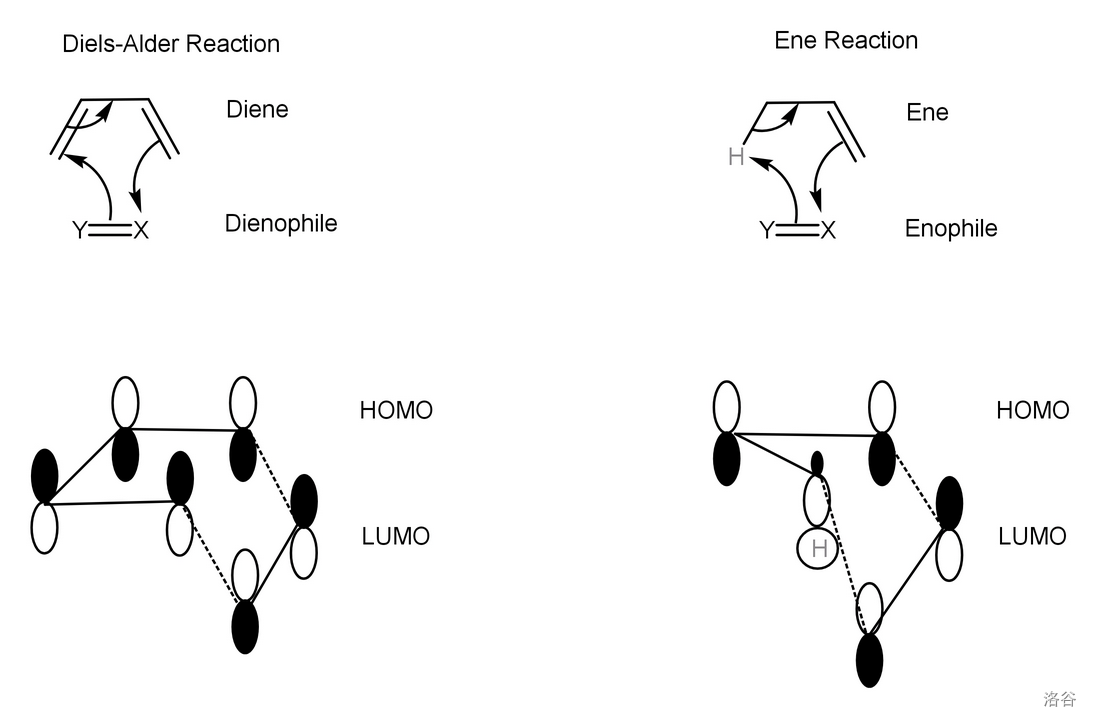
回到本反应。它实质上就是一个 烯反应 (Ene Reaction),只不过亲烯体(下图中的 $Y=X$)是单线态氧。烯反应的通式如下:$^{[25]}$
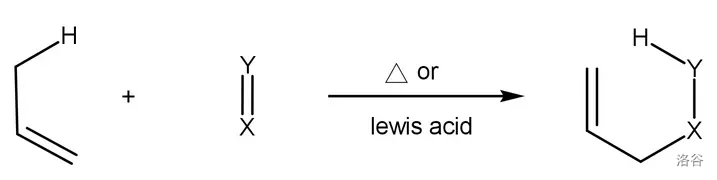
反应过程是亲烯体和烯的六中心 $σ$ 键迁移。烯烃的 $π$ 键、烯丙位的 $sp3-σC–H$ 键都参与周环反应。在此过程中双键的迁移、新的 $C–H$ 和 $C–C~σ$ 键的形成是协同进行的,与著名的 $Diels-Alder$ 反应类似:$^{[25]}$
本反应中,$4,5$ 位的烯烃先与单线态氧发生烯反应,得到过氧化物,随后让过氧化物在 $PMe_{3}$ 中分解,得到最后的产物:
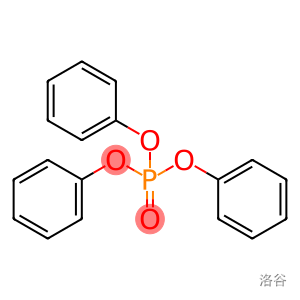
本反应中还使用了 $TPP$ 作为催化剂$^{[26]}$,其结构如下:
反应产率 $82\%$,且使用的反应物是廉价的氧气,能量源是光,其余试剂也是常见且廉价的,足以见到该反应的优越性。
3.12 第十二步
这一步可以分成五个小步。第一小步在之前已经出现过,就是用 $MsCl$ 把醇变成磺酸酯$^{[3]}$:
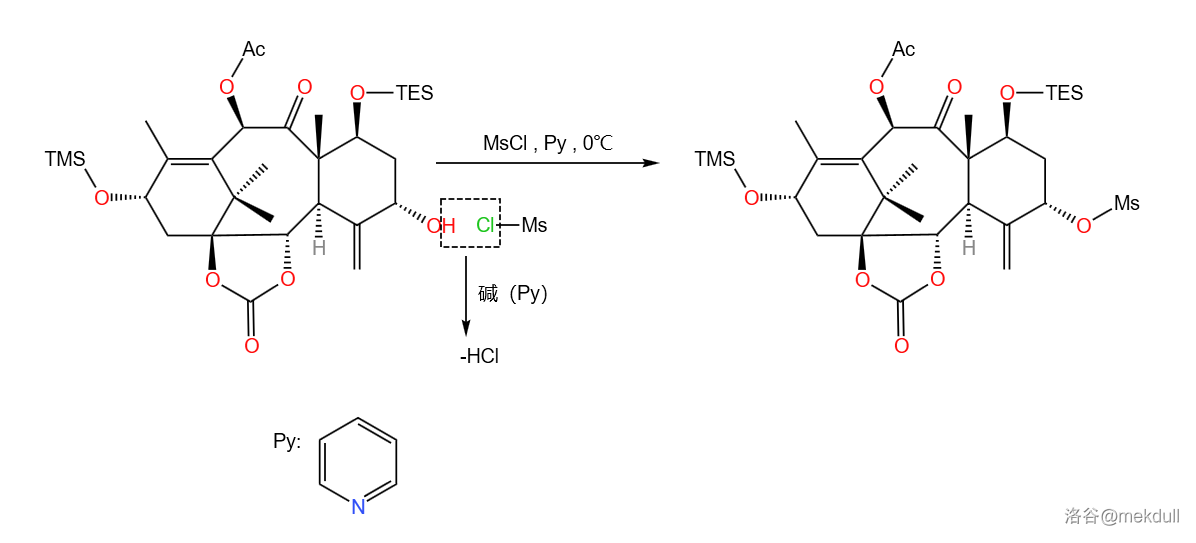
随后的第二小步也是我们非常熟悉的 $OsO_{4}$ 对烯烃的双羟化,之前出现的 $Lemieux-Johnson$ 反应中也有用到。反应如下图所示进行:$^{[29]}$
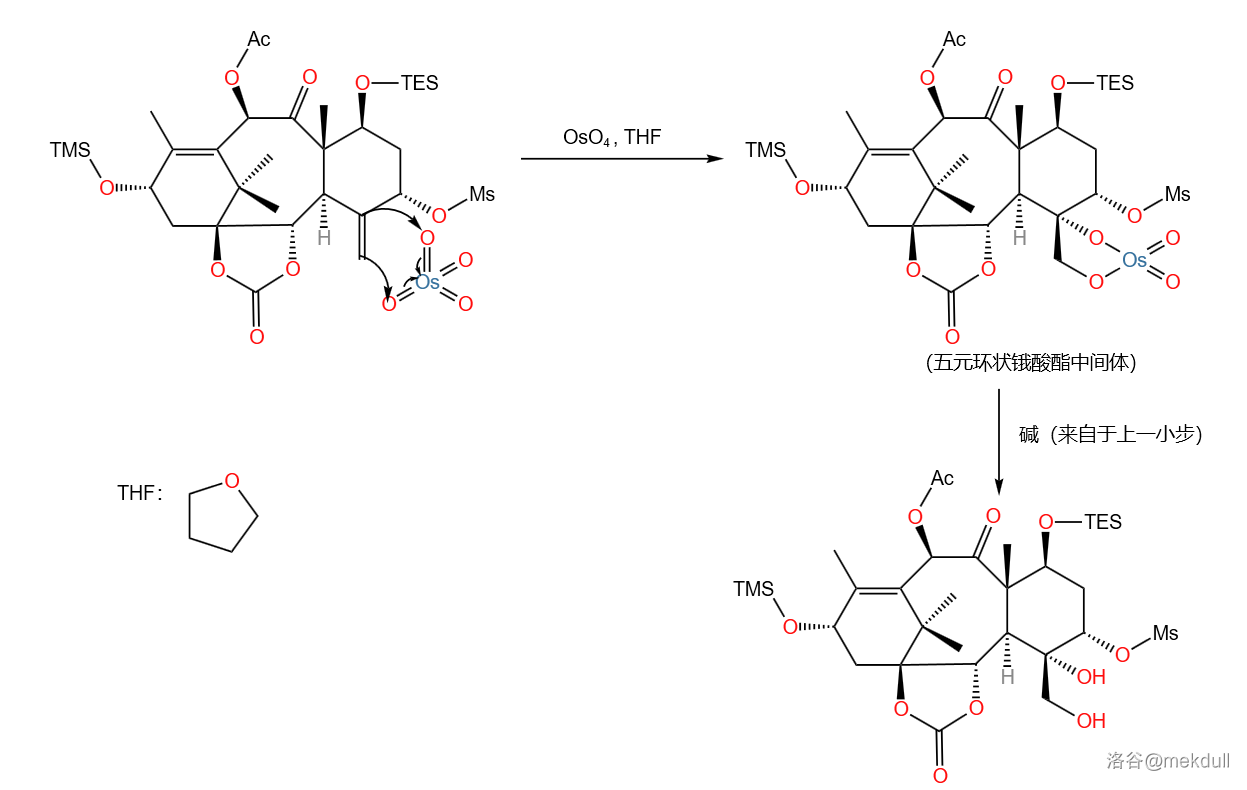
前两小步(实际应是一锅化的)产率为 $75\%$。
第三小步是一个变异的 $Williamson$ 醚合成反应,用于构筑氧杂环丁烷体系。这里使用碱兼催化剂的 $DIPEA$ $^{[29]}$ 脱去了 $MsOH$,使 $20$ 号位和 $5$ 号位成醚:
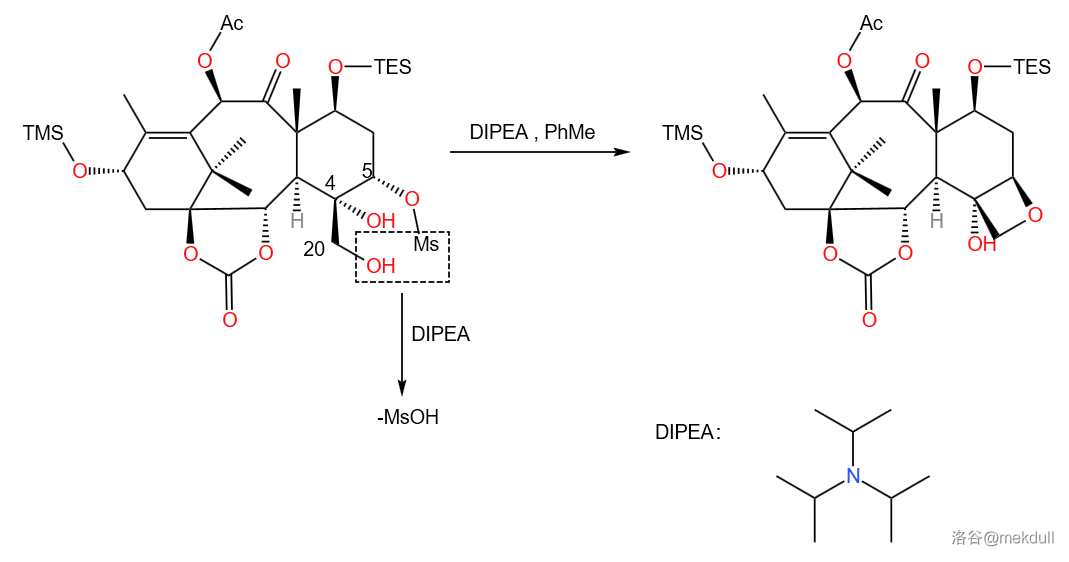
显然由于底物本身带有两个羟基,所以会出现 $5$ 号位不与 $20$ 号位成醚而与 $4$ 号位成醚得到的环氧化物副产物。但由于四元环比三元环稳定一些,所以实际上副产物产生是热力学不利的,故占比应该比主产物低许多。
第四小步即是使用 $Ac_{2}O$(乙酸酐)对 $4$ 号位上那个羟基进行乙酰化,使用的碱为之前出现过的 $DMAP$ $^{[10]}$:

最后,再顺便给 $13$ 位上的羟基脱个保护。使用的试剂是通用硅醚断裂试剂 $TBAF$(四丁基氟化铵):
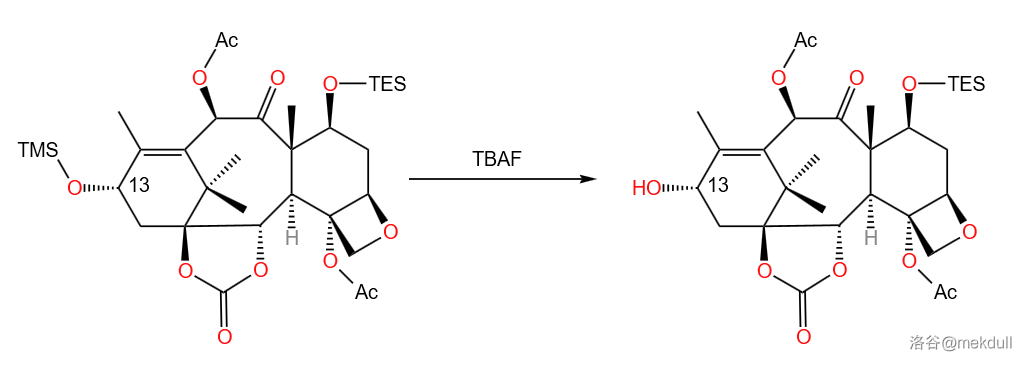
$TBAF$(四丁基氟化铵)是一种季铵盐,其结构如下$^{[30]}$:
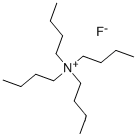
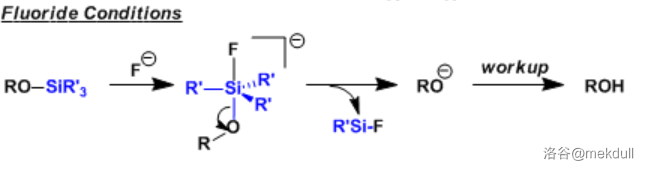
它脱去硅醚保护主要是靠在极性溶剂中释放出的 $F^{-}$ 离子。它作为亲核试剂可以进攻硅原子 $3d$ 缺电子的空轨道,形成五配位的中间体。由于 $Si-F$ 键键能很强,所以反应会向 $Si-O$ 键异裂生成氧负离子的方向进行:$^{[31]}$
可以观察到,在这步反应的主产物里,$7$ 号位上的 $TES$ 保护基没有被脱去,可能是由于空间受阻的原因。
后三步也应是一锅煮的,产率为 $32\%$。这一大步的总产率为 $24\%$。
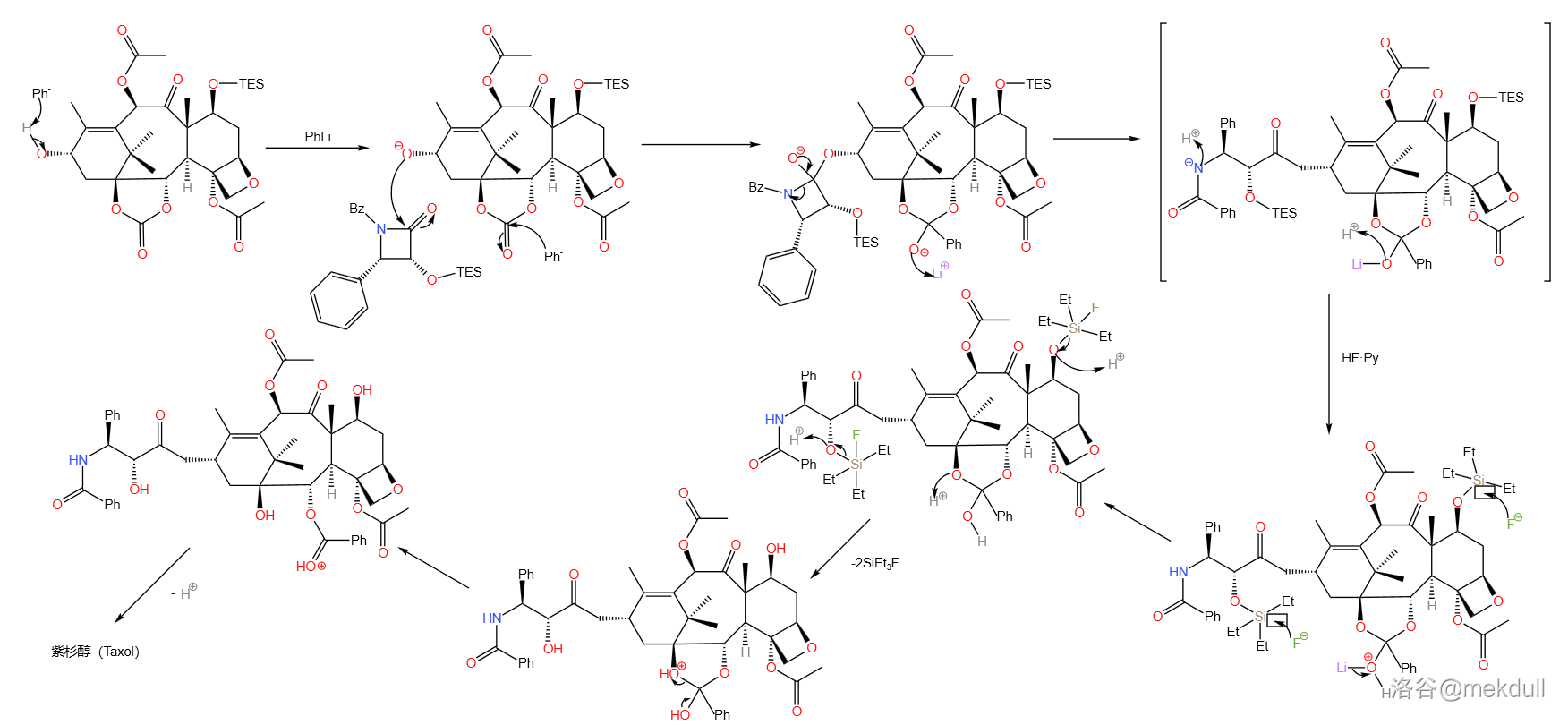
3.13 第十三步
这是整个全合成的最后一步,采用一锅煮的方式完成了 $13$ 号位的侧链安装、$2$ 号位的苯甲酸酯形成和所有硅类保护基的脱去。这种开创性的“三合一”反应极大的提高了反应效率与产率,也使反应步骤大幅缩减,集中体现了整个全合成体系的巧妙。反应机理如下:
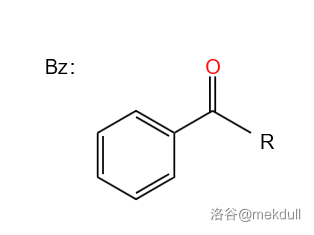
其中,$Bz$ 指苯甲酰基,结构如下:
首先使用强碱将 $13$ 号位上的羟基氢拔去,生成的氧负离子进攻另一底物的羰基,进而导致该底物不稳定的四元环开环,完成 $13$ 号位的侧链安装;同时,强碱进攻 $1,2$ 位碳酸酯的羰基,得到中间体(上图中用方框框出的物质);该中间体经过 $HF$ 处理,在脱去所有硅类保护基的同时在 $2$ 位生成苯甲酸酯,完成紫杉醇的全合成。
该反应的产率为 $68\%$。
4.总结
近半个世纪以来,得益于有机合成理论和分析技术的不断创新,天然产物全合成取得了长足发展。紫杉醇作为天然存在的抗癌物质,其人工全合成的意义不言而喻。李闯创团队报道的这例全合成是目前(截至 $2023$ 年年末)步数最少的全合成路线,总产率达到 $0.118\%$,平均每步产率达到 $70\%$ 以上, 将紫杉醇的全合成研究向前推进了一大步。
回首紫杉醇的研究历程,不禁令人感慨万千。 $1971$ 年,美国化学家 $Wani$ 及其团队在顶尖化学杂志《美国化学会志》($Journal~of~the~American~Chemical~Society$,简称 $JACS$)上报道了一项重磅研究成果,他们采用 $X$ 射线衍射等技术,确定了紫杉醇的结构。紫杉醇研究的大幕自此被拉开。过去五十几年,紫杉醇一直活跃在癌症的治疗中,并成为了最优秀的天然抗肿瘤药物之一。紫杉醇的天然来源 太平洋红豆杉 是世界公认的濒临灭绝的天然珍稀药用植物,其生长周期极为缓慢,从小树苗长成大树需要数百年。紫杉醇在太平洋红豆杉的树皮中的含量仅为 万分之一,但全球每年对紫杉醇的需求量却能达到吨级别。通常,治疗一个卵巢癌患者就需要 数棵百年以上树龄的太平洋红豆杉,这使得通过砍伐珍稀植物获取药物的做法不可持续,且 严重破坏生态环境。在这种情况下,研究紫杉醇的人工合成就是在挽救这种可怜的植物,挽救地球的生态。

(图:太平洋红豆杉)
紫杉醇的全合成仍在继续研究。目前的所有紫杉醇全合成路线都只停留在实验室中,而没有工业化,究其原因还是合成成本太高。因此,李闯创课题组的这例全合成是一块里程碑,但绝不是这项研究的终点。我相信,随着有机化学的发展,终有一天我们会拿下紫杉醇工业化合成的圣杯。
5.参考文献
$[1]$: 南科大李闯创团队JACS:完成紫杉醇的最短全合成
$[2]$: 文献精读 - 紫杉醇的全合成路线
$[3]$: 对甲苯磺酰氯的相关反应
$[4]$: OsO4/NaIO4氧化合成醛
$[5]$: 烯醇硅醚的制备合成 何佳乐
$[6]$: Prilezhaev反应
$[7]$: 硫代缩醛(酮)法
$[8]$: 妙不可言的化学——三光气在有机合成中的妙用
$[9]$: 频哪醇偶联反应
$[10]$: 4-二甲氨基吡啶(DMAP)
$[11]$: DMAP及其类似物
$[12]$: 常用氧化剂——四丙基高钌酸铵 (TPAP)
$[13]$: TPAP(Ley-Griffith)氧化反应
$[14]$: 苄基保护基
$[15]$: 三乙基硅基三氟甲磺酸酯
$[16]$: 醇的硅醚保护反应 Silyl Protective Group
$[17]$: 常用氧化剂——氯铬酸吡啶(PCC)
$[18]$: 对甲苯磺酰肼
$[19]$: 通过烯丙基二氮烯重排进行非环1,4-立体控制:甲苯磺酰Formation形成中动力学E立体选择性的发展,应用和基本作用
$[20]$: 三甲基硅咪唑
$[21]$: 三甲基硅咪唑(TMSim)
$[22]$: 儿萘酚硼烷
$[23]$: 单态氧
$[24]$: 活性氧
$[25]$: 有机人名反应——Ene反应(Alder-ene反应、烯反应)
$[26]$: 磷酸三苯酯
$[27]$: Singlet Oxygen Ene Reaction - Harvard University:单线态氧烯反应
$[28]$: Caglioti 反应
$[29]$: N,N-二异丙基乙胺
$[30]$: 四丁基氟化铵
$[31]$: 醇的硅醚保护反应 Silyl Protective Group