谨由此文,纪念世界有机合成泰斗岸义人($Yoshito~Kishi$)教授逝世一周年;纪念岩沙海葵毒素全合成被人类完成三十年。
本文献给岸义人教授,献给所有化学工作者,还有每一个热爱化学的人。
注:如果本文中有任何错误或疏漏,请务必联系作者或在评论中写出。不胜感谢!
1.前情提要
岩沙海葵毒素($palytoxin$,简称 $PTX$),亦称沙海葵毒素或群体海葵毒素,是一种提取自毒沙群海葵(刺胞动物门,珊瑚纲,六放珊瑚亚纲,海葵目,皮沙海葵科,沙群海葵属)的非蛋白毒素,也是已知非蛋白毒素中毒性最强烈的毒素之一。软珊瑚、玫瑰海葵等多种海洋有毒生物中都含有这种物质。
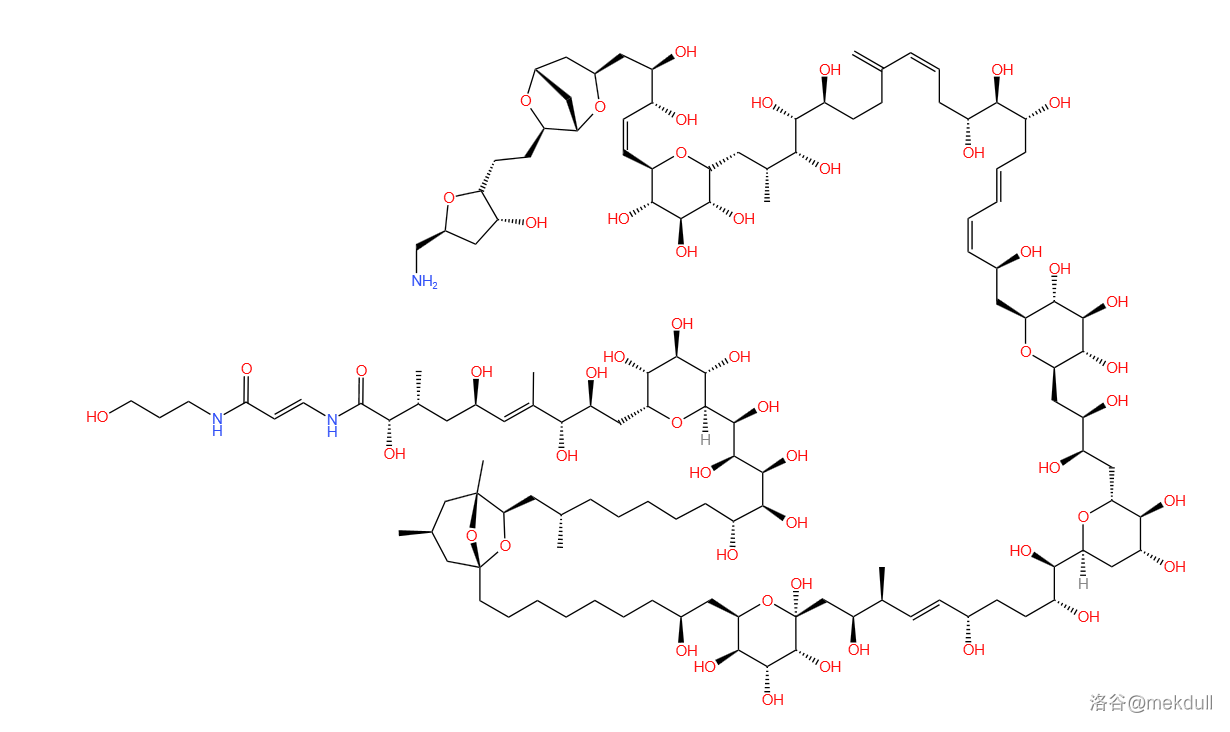
(上图:岩沙海葵毒素的结构式)

(上图:发表于 $Science$ 上的图片,为现实中的毒沙群海葵)
其分子式为 $C_{129}H_{223}O_{54}N_{3}$,分子量高达 $2680.2$。它有着巨大而复杂的结构,并拥有 $64$ 个手性碳原子、$42$ 个羟基、$1$ 个五元环醚、$5$ 个六元环醚、$2$ 个七元桥环双醚和 $2$ 个特殊的酰胺结构(即酰烯胺结构),这无疑为它的全合成工作带来了巨大的挑战。因此,它也被誉为“有机合成界的珠穆朗玛峰”。
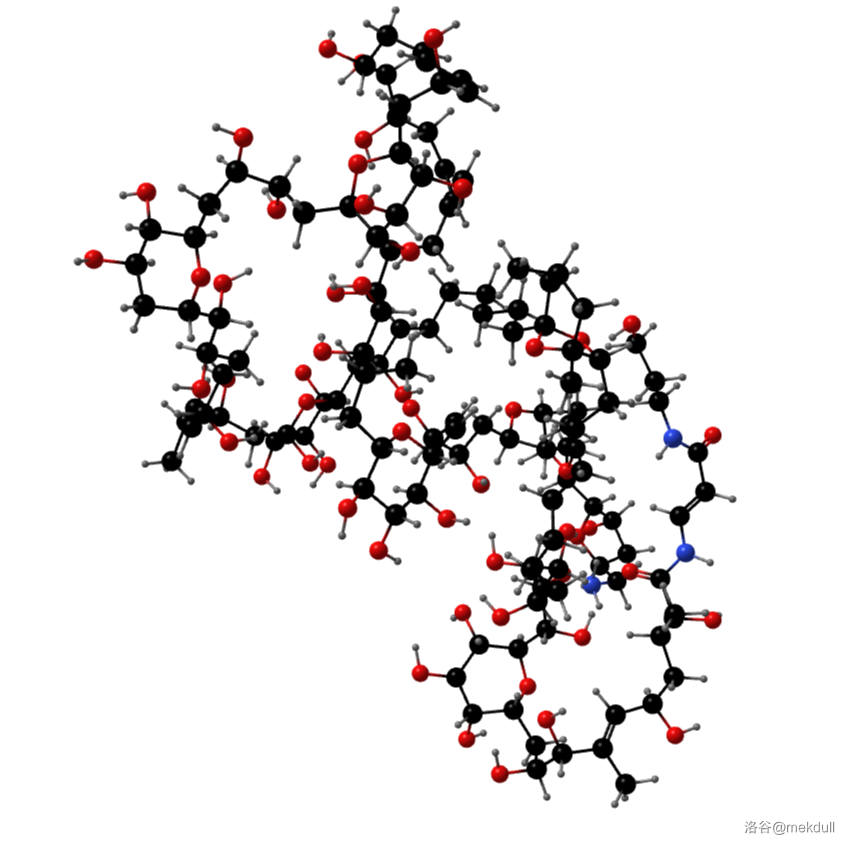
(上图:计算机 $3D$ 渲染得到的岩沙海葵毒素的立体结构)
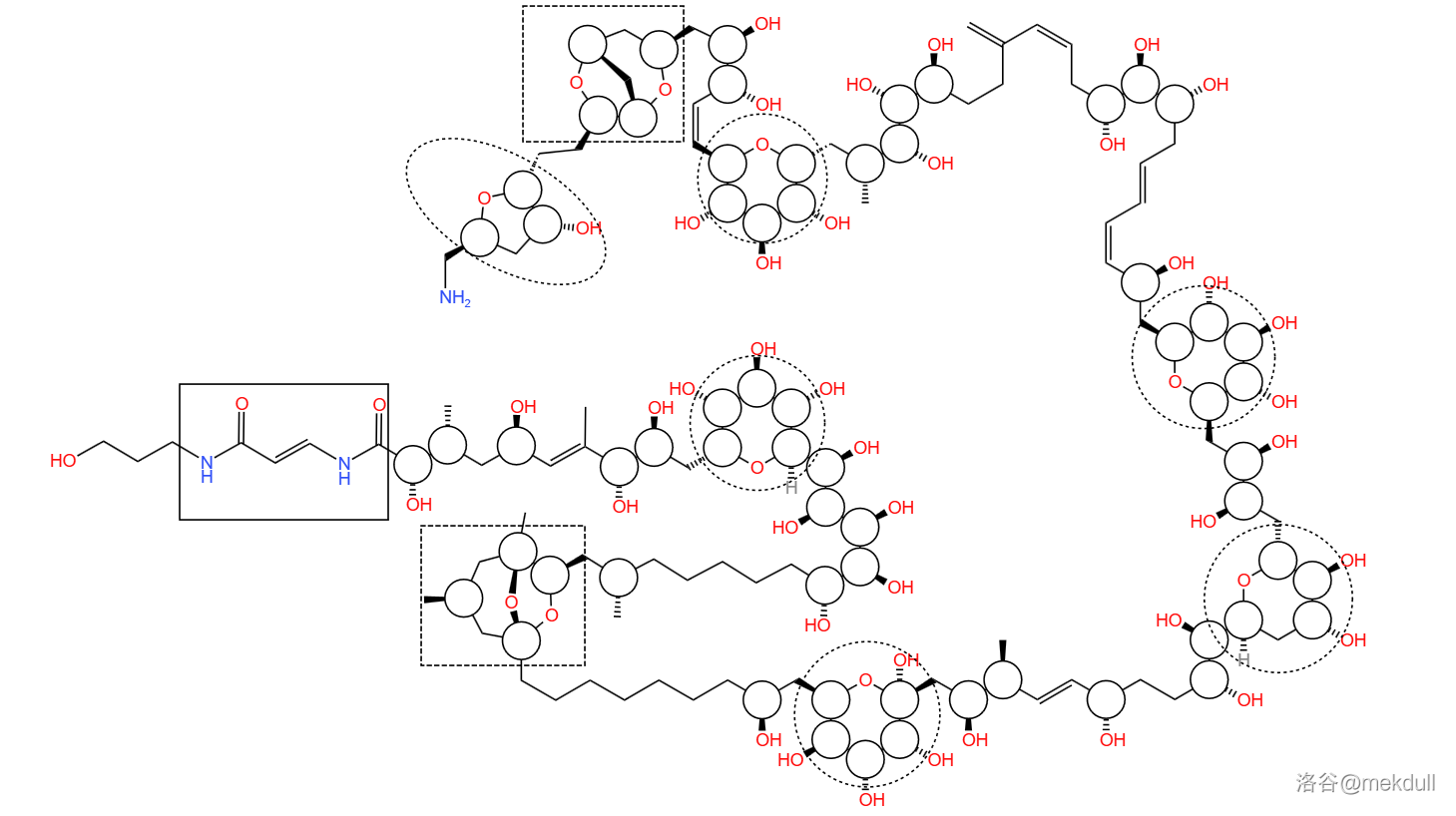
(上图:使用特殊标识表示了岩沙海葵毒素中的特殊结构,小实线圆表示手性碳原子,大虚线圆为 $6$ 元环醚结构,大虚线椭圆为 $5$ 元环醚结构,虚线方框框出了 $7$ 元桥环双醚结构,实线方框框出了两个特殊酰胺,即酰烯胺结构)
但是,人类不会因此放弃。
事实上,自从 $1971$ 年 $Sheuer$ 等人从海藻类珊瑚中分离出它之后,岩沙海葵毒素便成为了有机化学界的焦点。$1981$ 年,$Moore$ 课题组和 $Daisuke~Uemura$ 课题组独立报道了它的平面结构,并在第二年完成了其立体结构的表征。随即,许多有机合成化学家便开始攀登全合成的高峰。这一难题最终在 $1994$ 年被哈佛大学的日裔化学家岸义人 ($Yoshito~Kishi$) 带领的课题组解决,前后耗时 $12$ 年。在下文中,我们将详细讨论和解析这条全合成路线。
2.概述
岩沙海葵毒素事实上是一个非常线性的分子,因此采用的全合成思路和环系的紫杉醇等有很大不同。岸义人课题组采用的合成策略是砌块拼接法,即现将分子拆分成几个较小的砌块,再将它们拼接起来。听起来这种方法相当的原始、暴力,但却是合成线性分子最常用的手段,而且还有利于使用不同的保护基保护分子中大量的羟基。课题组拆分出的砌块结构如下,一共 $9$ 个:
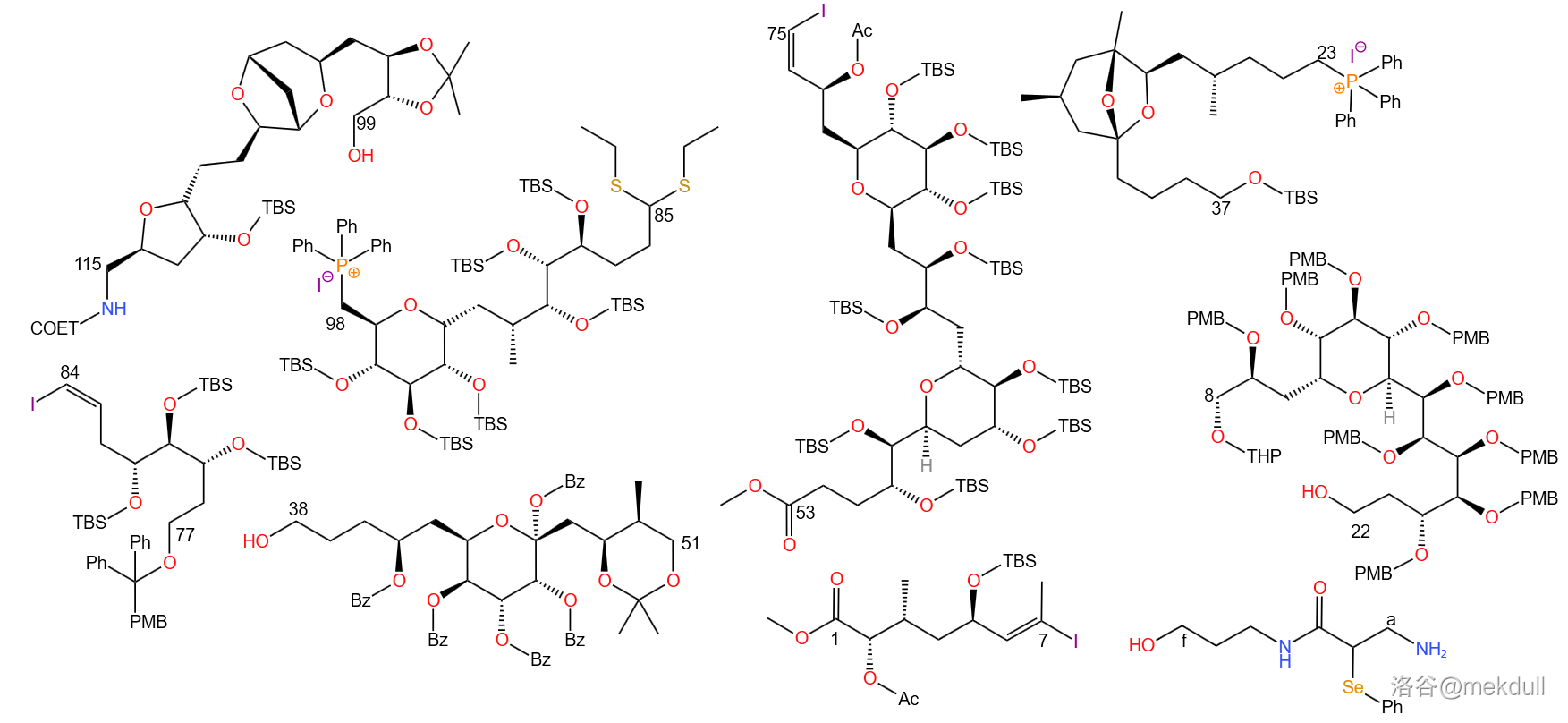
(上图:岩沙海葵毒素全合成中,使用的 $9$ 个合成砌块)
这 $9$ 个砌块不是随便划分的,每个砌块的端基都尽量选择无手性碳原子,这样可以减少合成过程中手性中心形成的困难。连接这些砌块所需的即是偶联反应。
在 $1985$ 年,即课题组开始研究全合成的第三年,他们就已经完成其中 $8$ 个砌块的合成(除去右下角的那个砌块)。$1989$ 年,课题组将这 $8$ 个砌块拼接,得到的产物名为岩沙海葵毒素羧酸,是合成岩沙海葵毒素的重要前体。

(上图:岩沙海葵毒素羧酸的结构,相比岩沙海葵毒素,它在一端少了几个碳原子,因为还有一个砌块没有接上去)
$5$ 年后,$1994$ 年,课题组终于将最后一个砌块拼到了岩沙海葵毒素羧酸上,完成了全合成。接下来,我们重点讨论的就是怎么把这几个砌块拼起来。(对于这几个砌块怎么合成,由于篇幅和资料原因,在此姑且不表)
本文中涉及到的主要有机反应清单:
-
$Nozaki-Hiyama-Kishi$ 反应;
-
$Suzuki-Miyaura$ 偶联反应,$TlOH$ 改进法;
-
$Wittig$ 反应;
-
$Matteson$ 硼酸酯化与增碳反应;
-
$Hoener-Wadsworth-Emmons$ 反应,一种改进的 $Wittig$ 反应;
-
$Swern$ 氧化;
-
$Luche$ 不对称还原;
-
$Davis$ 氧化。
3.逐步合成分析
为了方便讲解,我们给上述 $9$ 个砌块编号。最右下角那个我们令它为 $9$ 号砌块(因为它最晚被拼接上去),其他的砌块按照其链首碳原子的编号排序,如下图所示。同时,我们把分子分为左右两大部分,分别完成这两部分的拼接之后再把它们连接起来,最后脱去保护基,再连上 $9$ 号砌块完成全合成。

接下来,请坐好抓稳,全合成的介绍即将正式开始。
3.1 左半部分的合成
首先,$2$ 号砌块在 $DMSO$ 与草酰氯的作用下,发生 Swern氧化反应,随即使用碱($Et_{3}N$)淬灭。这一步将 $22$ 号碳原子转变为醛羰基:

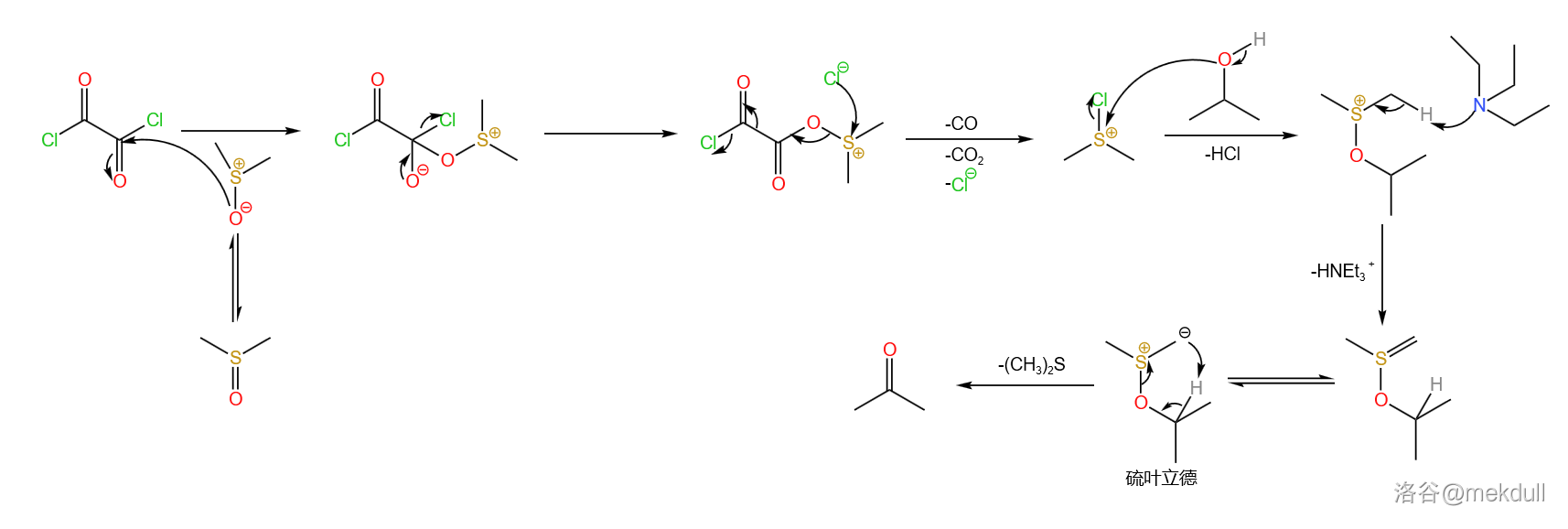
Swern氧化是目前最有选择性的将醇氧化为醛或酮的方法之一,被广泛用于复杂天然产物的合成。其反应机理如下,经过硫叶立德中间体:(反应机理以异丙醇实例,其余醇同理)
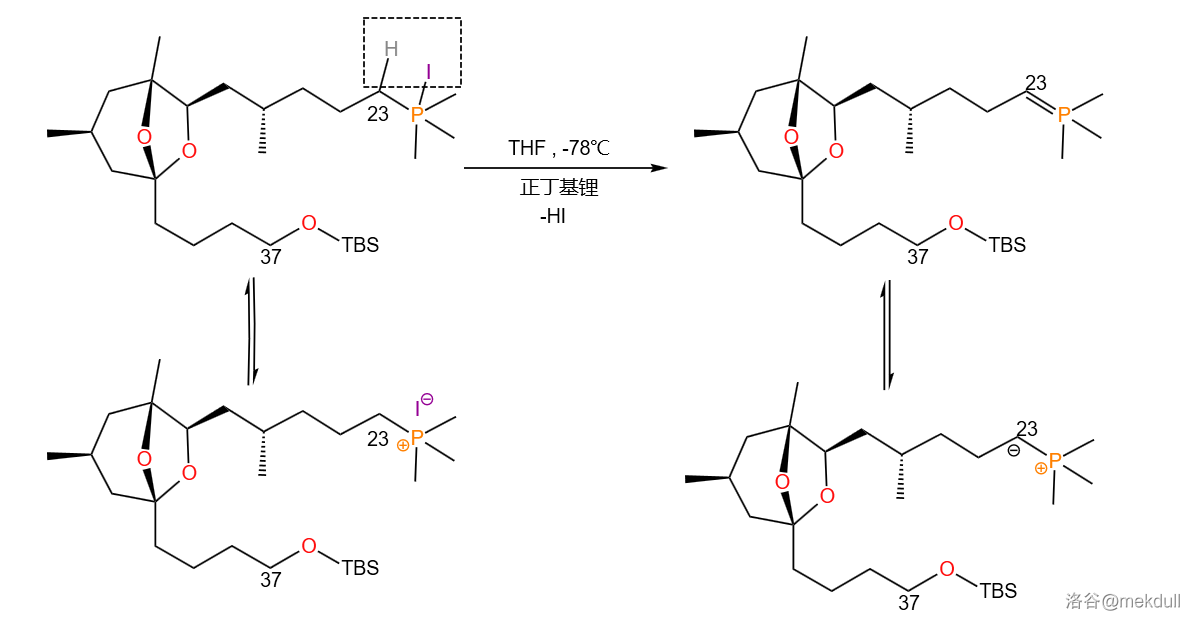
随后,砌块 $3$ 在低温($-78^{\circ}C$)的 $THF$ 中被强碱(正丁基锂,$^{n}BuLi$)消去 $1$ 分子 $HI$,得到磷叶立德:(注:与 $P$ 原子相连的三个苯基在这里被省略了)
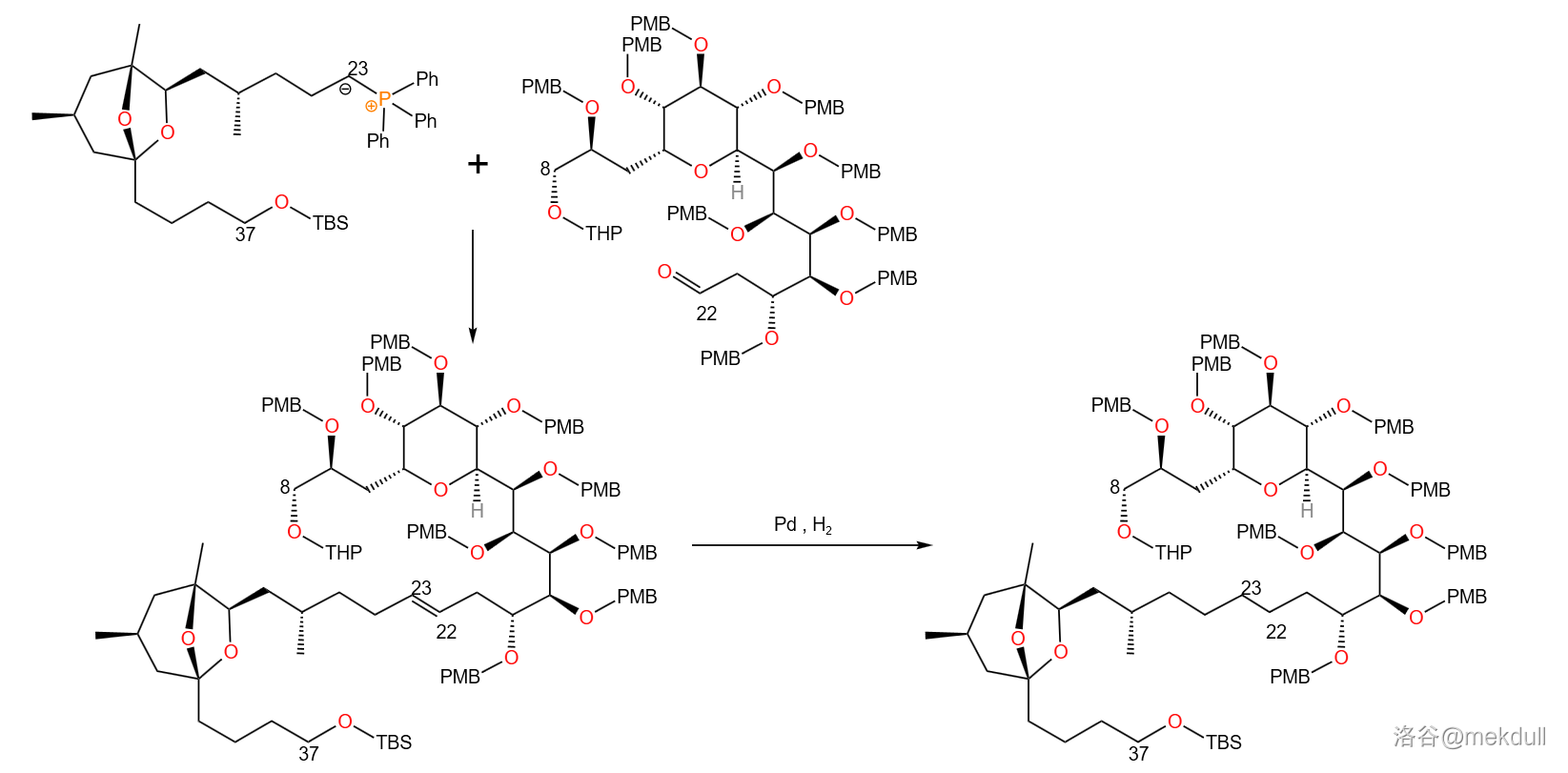
接着便是两个砌块之间发生 $Wittig$ 反应,生成 $22$ 与 $23$ 号碳原子之间的双键,将两个砌块拼在一起。随即使用 $Pd$ 对其催化加氢,将 $22$ 与 $23$ 号碳原子之间的键变成单键:
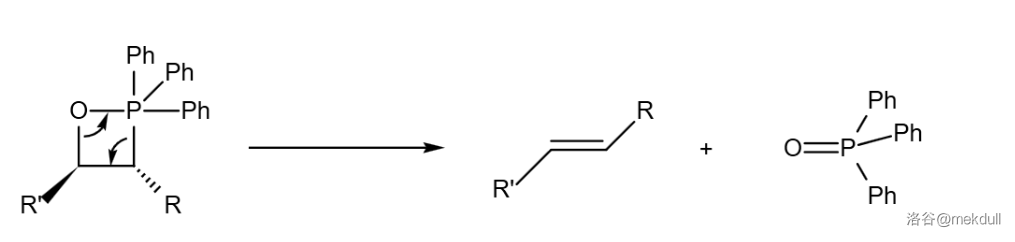
$Wittig$ 反应的机理一般认为是磷叶立德的碳负离子进攻羰基,随后环化形成氧磷杂环丁烷中间体。整个过程也可以看成一个 $[2+2]$ 环加成:

随后四元环碎裂,消去氧化三苯基膦($(Ph)_{3}P\text{=}O$)后得到烯烃。在整个反应过程中,键能强的磷氧键形成是主要推动力。
言归正传,反应后得到的产物随后在常温的 $THF$ 中被 $TBAF$(四丁基氟化铵)脱去 $TBS$ 保护基,在 $37$ 号位得到游离的醇羟基:

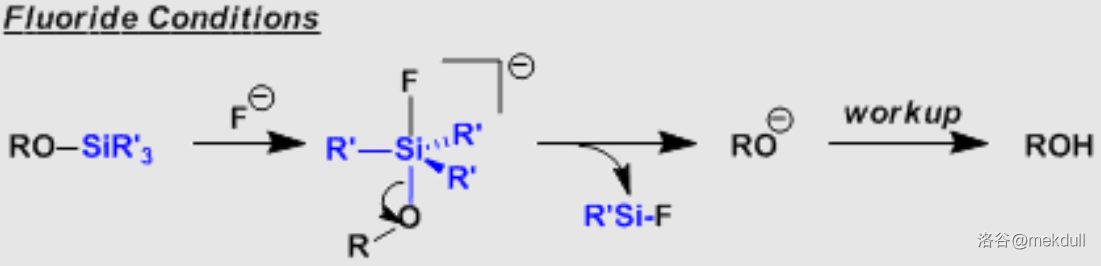
$TBAF$ 作为脱去硅类保护基的通用试剂,脱保护的机理一般认为经过五配位中间体,最后一步 $Si-O$ 键断裂的动力来源于键能更强的 $Si-F$ 键的生成:
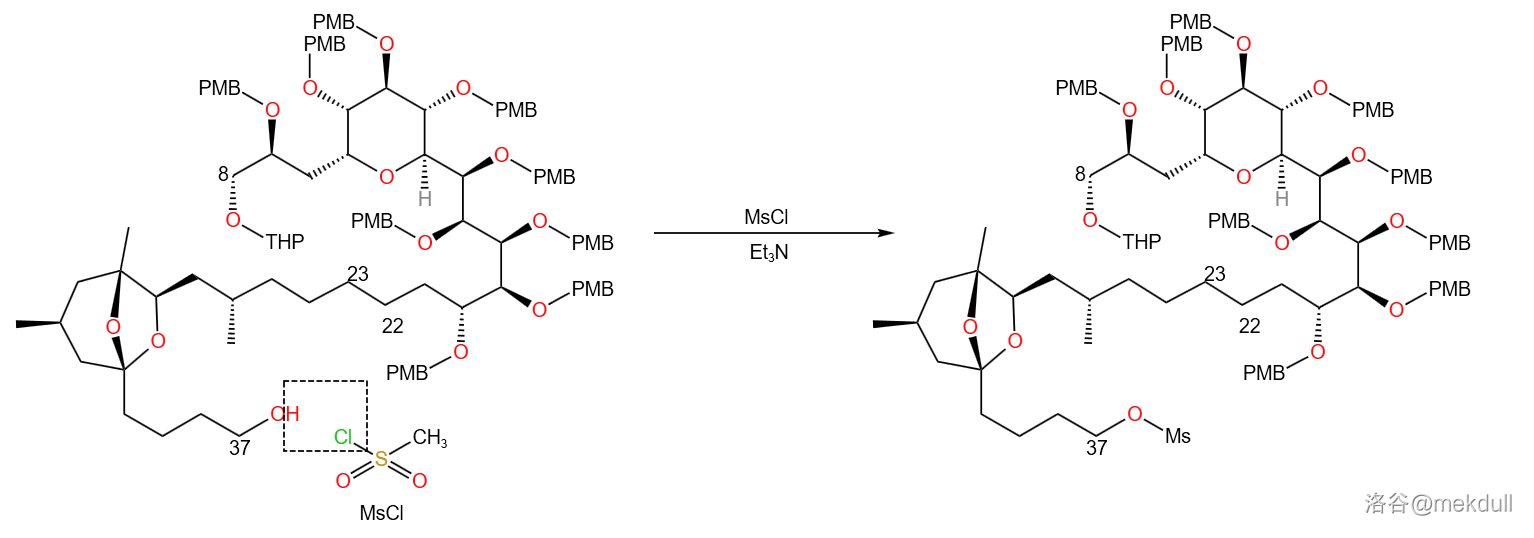
下一步便是在冰浴中,$37$ 号位上的醇羟基与 $MsCl$(甲磺酰氯)反应得到甲磺酸酯。反应同时会产生酸,因此使用了三乙胺($(C_{2}H_{5})_{3}N$)作为缚酸剂,防止体系随着反应进行而不断酸化从而导致条件改变、产率降低:
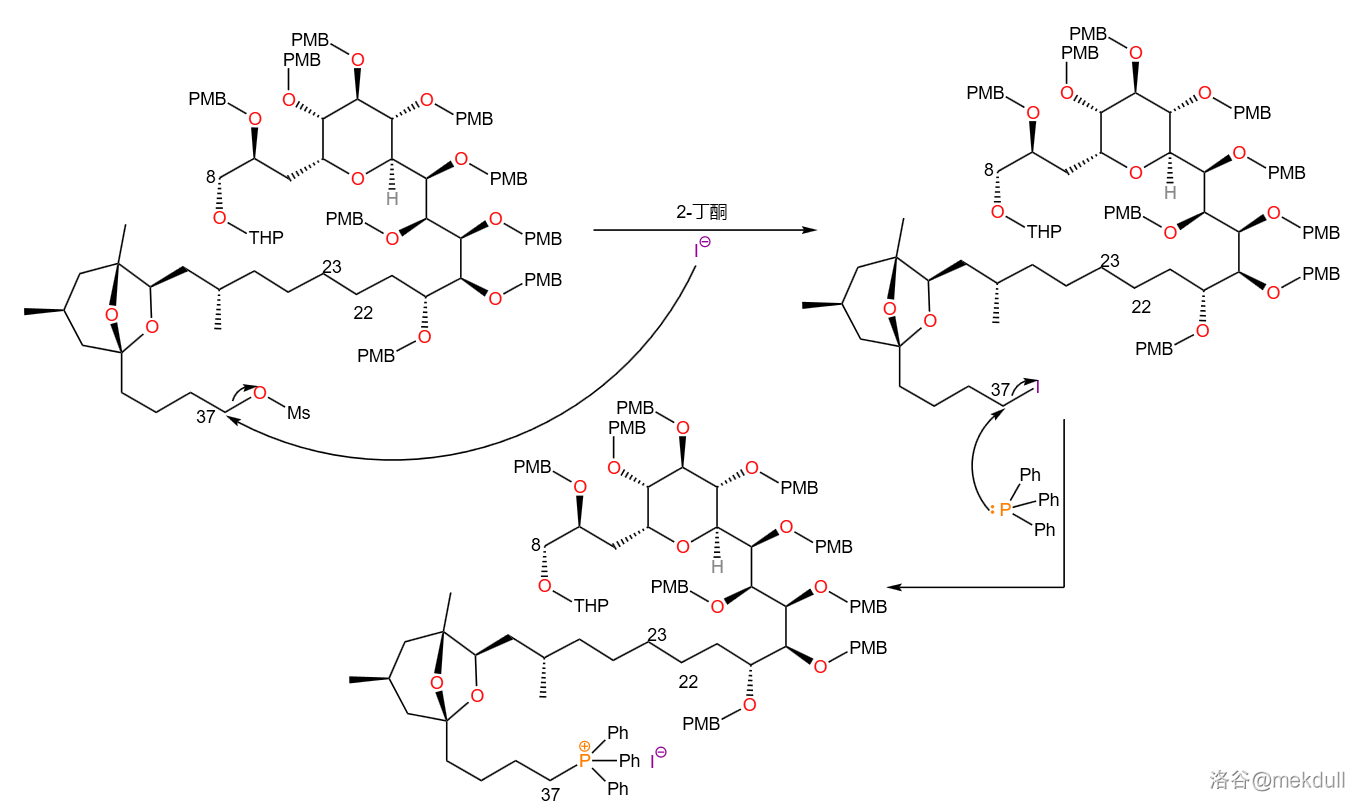
甲磺酸酯是有机合成中常用的易离去基团,所以下一步就是让它在 $2-$丁酮溶剂中与碘化钠($NaI$)反应,甲磺酸酯基离去得到碘代烃。随后碘代烃与 $PPh_{3}$ 反应,得到鏻盐:
接下来便是重复一遍上面的反应程序:鏻盐被正丁基锂($^{n}BuLi$)拔去得到磷叶立德;砌块 $4$ 发生 $Swern$ 氧化得到 $38$ 号位上的醛;随后两者发生 $Wittig$ 反应得到 $37,38$ 号碳原子之间的碳碳双键;最后用 $Pd$ 进行催化氢化,完成 $2,3,4$ 三个砌块的连接:

接下来就是 $2$ 号砌块。与前面不同的是,这个砌块的拼接需要考虑 $8$ 号碳原子上的手性问题。事实上,砌块的划分已经尽量避免了手性合成步骤,但奈何目标分子的手性碳原子实在太多,不可能完全逃过手性这一关。
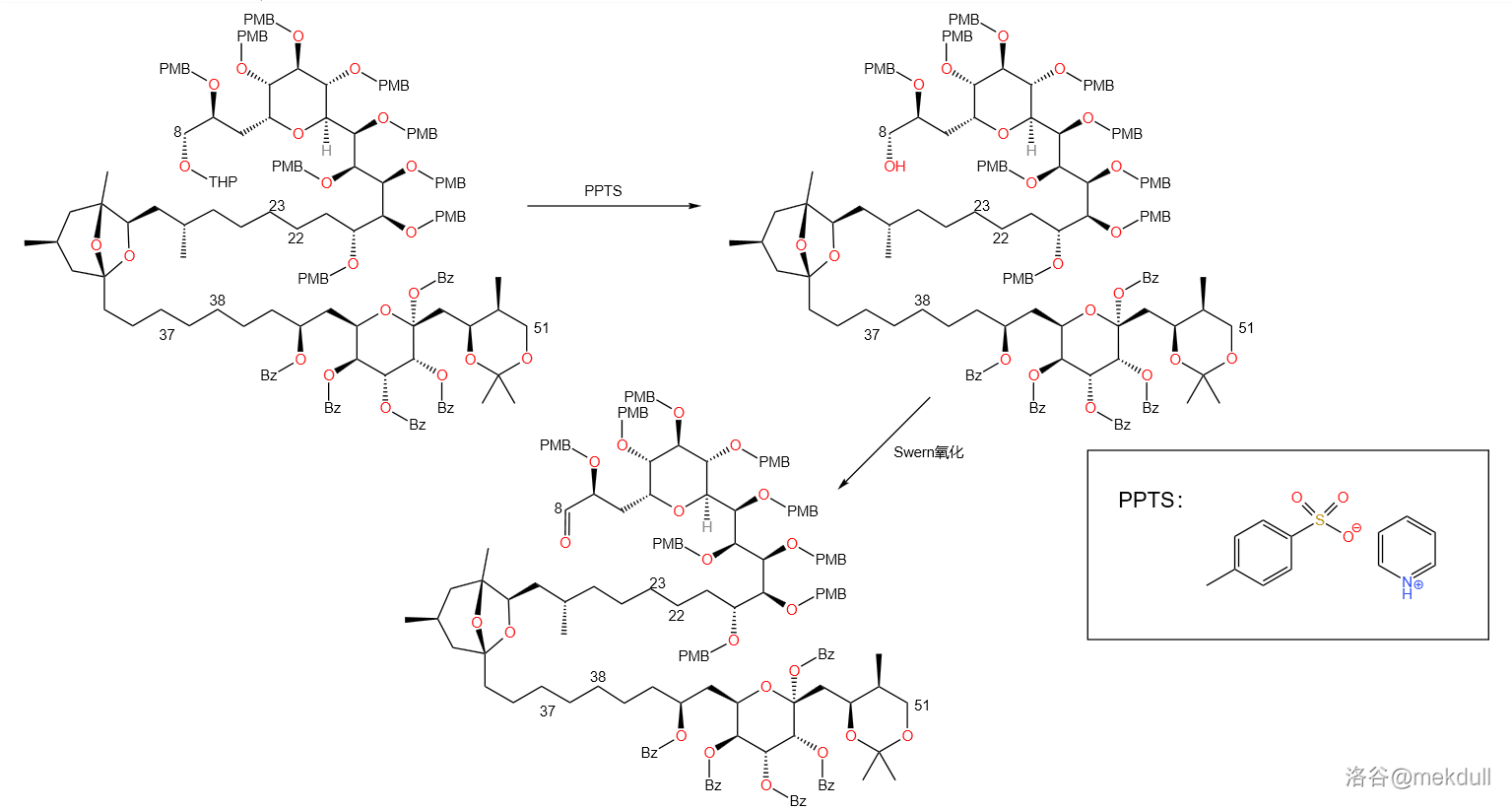
操作的第一步仍然是脱去 $8$ 号碳原子上的 $THP$(四氢吡喃)保护基,这一步使用了 $PPTS$(对甲苯磺酸吡啶盐)。相比于其他酸性条件脱保护的试剂,$PPTS$ 更加温和,故而具有两个明显的优点:
-
使得 $8$ 号碳原子上的 $THP$ 保护基与 $51$ 号碳原子上的缩丙酮保护基活性差异放大。如果使用更活泼的试剂,那么 $51$ 号碳原子上的缩丙酮保护也有可能被脱去,但 $PPTS$ 则会高选择性地优先与 $8$ 号碳原子上的 $THP$ 基团反应;
-
由于底物分子对酸较为敏感,使用酸性试剂可能破坏分子结构,产生副产物并使产率降低。
游离出醇羟基后,下一步仍然是 $Swern$ 氧化将 $8$ 号位氧化为醛羰基:
紧接着的即是 $Nozaki-Hiyama-Kishi$ 反应(下文中统一简称 $NHK$ 反应)。在 $Cr(\text{II}),Ni(\text{II})$ 物种的共同催化下,烯基碘化物($1$ 号砌块)与醛之间发生偶联反应,得到 $\alpha,\beta$ 不饱和醇:
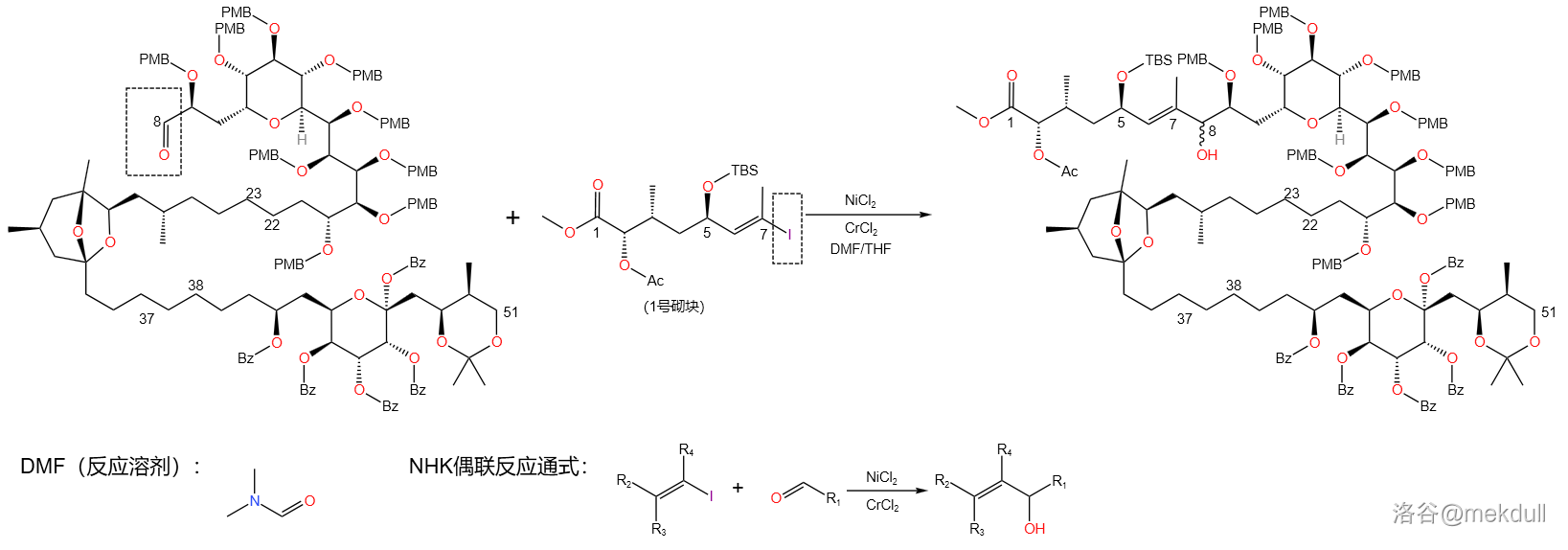
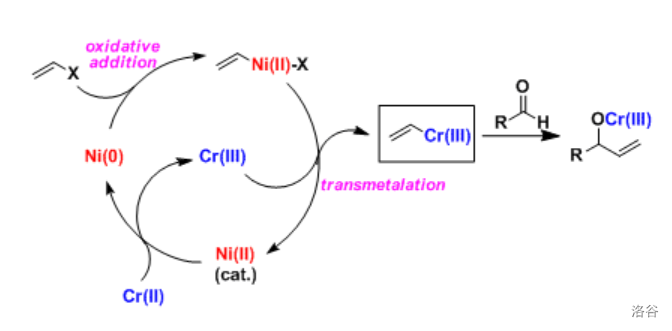
$NHK$ 反应机理如下,经历一个转金属化过程。反应的实质是烯基金属化合物中的碳负离子进攻羰基,得到的产物水解之后生成醇。
碳负离子进攻羰基是有方向性的,如果是醛羰基的话,则一般倾向于从位阻较小的氢原子一侧进攻($Cram$ 规则)。经过研究,课题组发现 $5$ 号碳原子上羟基的保护基居然对产物的构型比有很大的影响,应该是这个保护基影响了整体的空间位阻。于是,课题组开始对保护基进行筛选,最后发现:使用 $TBS$ 保护基时,所需构型与其非对映异构体比例为 $5:1$。
看起来已经挺不错了?但对于课题组来说,这还不够。于是,课题组采用了先氧化后还原的策略,将几乎全部的产物转化为了所需构型。首先仍然是 $Swern$ 氧化,紧接着是一个不对称的 $Luche$ 还原:
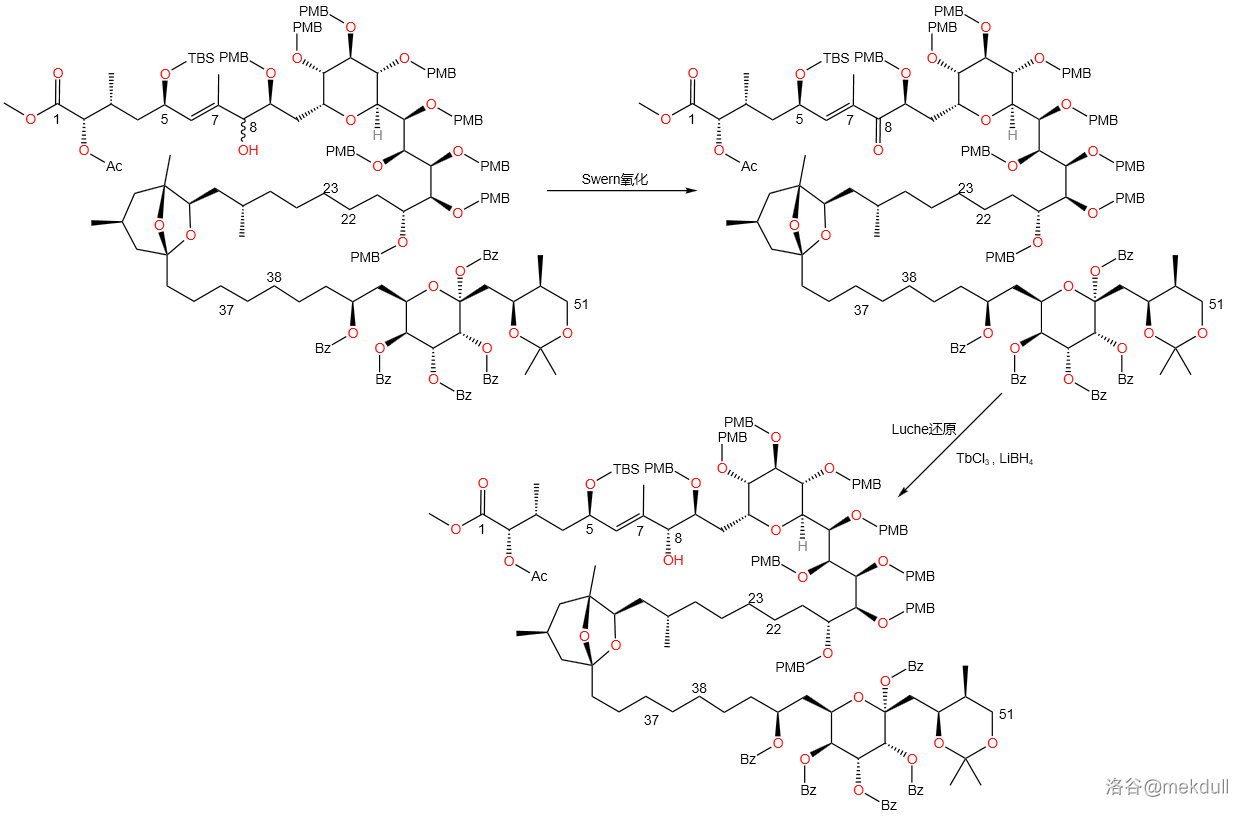
(此处 $Luche$ 还原应该还需要一个醇类物质)
为了找到还原反应最好的催化剂,课题组直接对稀土元素盐酸盐进行了筛选,最后发现 $TbCl_{3}$(三氯化铽)效果最好,结合筛选出来的 $LiBH_{4}$ 作为还原剂,可以把产物中非对映异构体的比例缩小到聊胜于无。
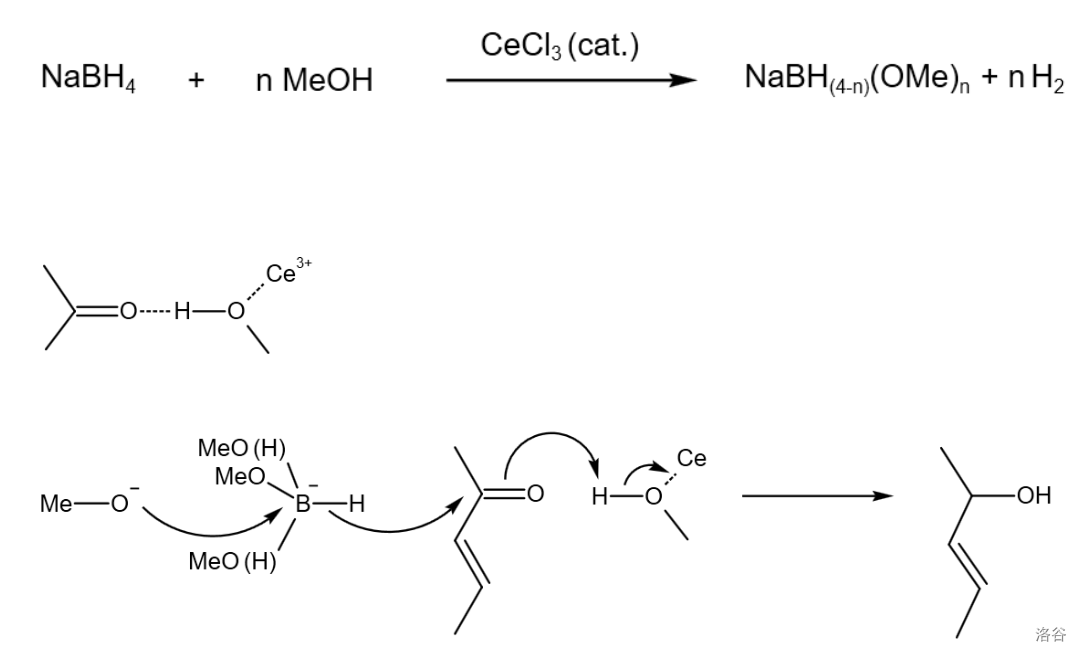
$Luche$ 还原本质上是氢负离子的还原反应,而稀土元素离子作为路易斯酸,通过配位不仅使得构型具有选择性,而且增加了醇的活性,也提高了羰基的亲电性,从而达到催化效果。机理如下:
这样一来,左半部分的四个砌块($1,2,3,4$ 四个砌块)已经被我们拼接到一起了。剩下的便是为之后两个砌块的拼接做准备。先用乙酸酐($Ac_{2}O$)保护 $8$ 号位上的羟基,然后再用 $PPTS$ 在较强烈的条件下脱去 $51$ 号碳原子上的缩丙酮保护,最后使用 $\ce{Ru(PPh3)3Cl2}$ 对 $51$ 号碳上的伯醇羟基选择性氧化:

$\ce{Ru(PPh3)3Cl2}$ 是一个常用的选择性氧化剂,其中钌元素氧化态为 $+\text{II}$ 。由于位阻超大,它只能氧化伯醇而不能氧化仲醇。至此,课题组完成了左半部分的全合成。
3.2 右半部分的合成
这一部分的合成比较复杂,也出现了很多原本在课题组“意料之外”的难题。但也正是这些难题,让它更为精彩。
首先,$8$ 号砌块发生 $Swern$ 氧化,得到 $99$ 号位的醛羰基,这一步并没有什么意料之外的事:
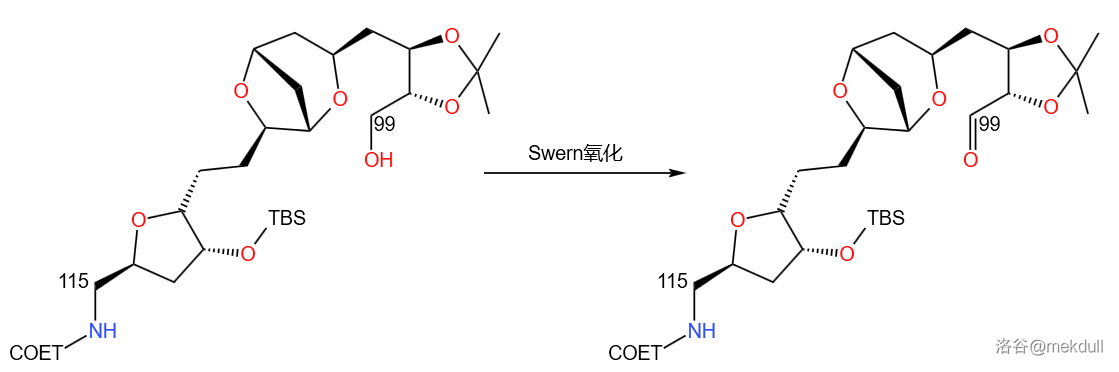
接着是将 $7$ 号砌块消去 $1$ 分子 $HI$ 得到磷叶立德,选择了 $HMPA$(六甲基磷酰胺,$\ce{((CH3)2N)3P=O}$)与 $THF$ 一起作为溶剂,$LDA$ 作为碱性试剂。这步进行得也是四平八稳:

但下一步就出事了。在两个砌块发生 $Wittig$ 反应时,课题组发现 $97$ 号碳原子会发生差向异构化,具体历程是先开环后关环。这样一来,$97$ 号碳原子的构型就变了。如果不注意这个小细节,那么后果便是满盘皆输。所幸课题组临危不乱,使用筛选溶剂大法成功解决了这个问题,没有因此而影响整体合成路线。
差向异构化:含有两个或以上手性中心的化合物分子中某手性中心的构型通过化学反应转换成其相反构型的过程。(摘自百度百科)
刚才的坏消息解决了之后,课题组发现其实还有一个好消息:这个反应的顺反选择性良好,顺反比直接就达到了令人满意的 $8:1$。于是课题组便没有继续优化,而是直接分离了两种产物。

得到所需产物后,下一步便是将 $85$ 号碳原子处的缩硫醇保护脱去。课题组采用了氧化脱去的方法,使用 $I_{2}$ 作为氧化剂,$Na_{2}CO_{3}$ 提供弱碱性环境:
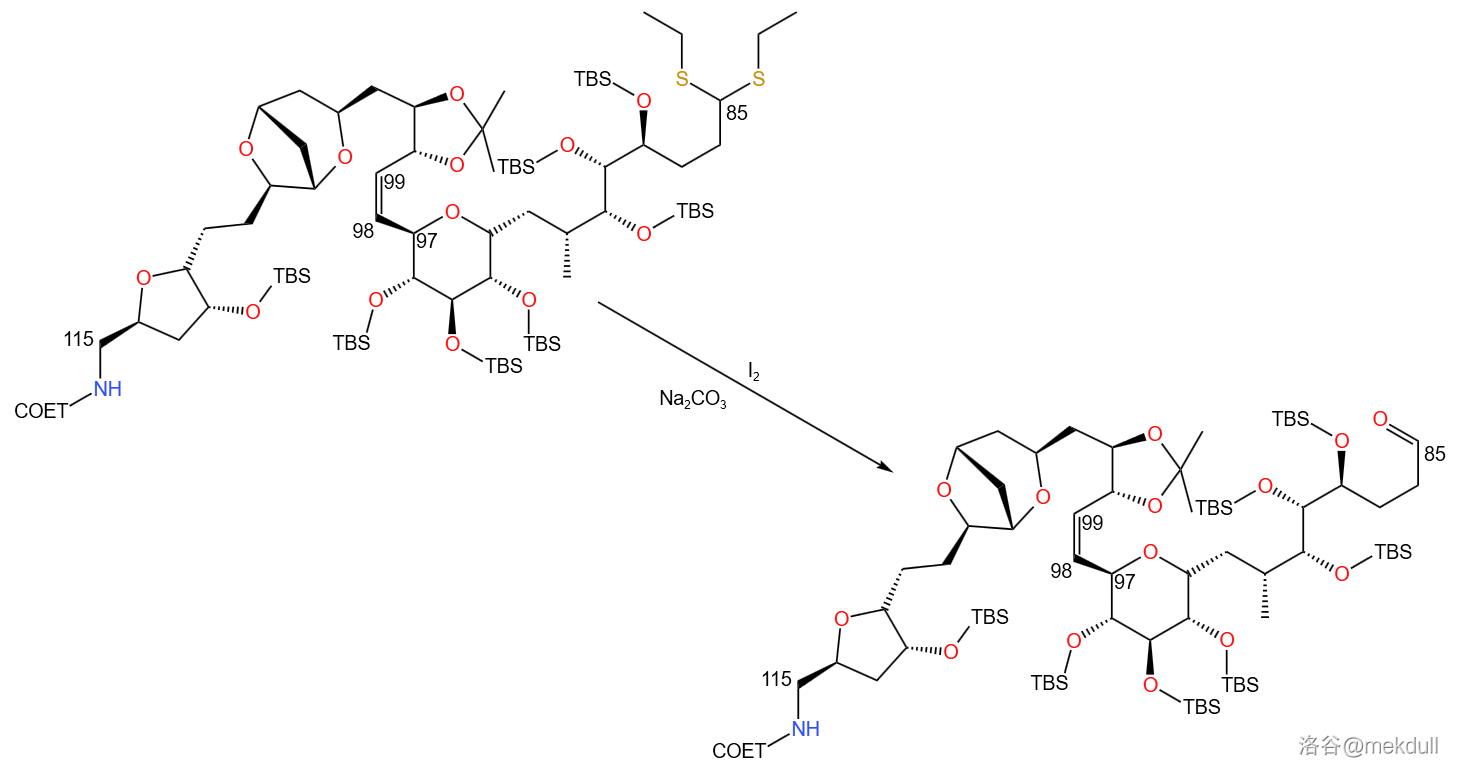
接着,与 $6$ 号砌块之间发生 $NHK$ 偶联反应,得到 $85$ 号位的醇羟基。此时 $85$ 号碳原子构型并不单一,但不需要额外进行什么操作,因为下一步就会用 $PDC$ 将 $85$ 号位氧化为酮羰基,进而和亚甲基三苯基膦($CH_{2}PPh_{3}$,其实就是由碘甲烷($CH_{3}I$)制备的磷叶立德)进行亚甲基化反应,得到 $85$ 号碳原子上的亚甲基:
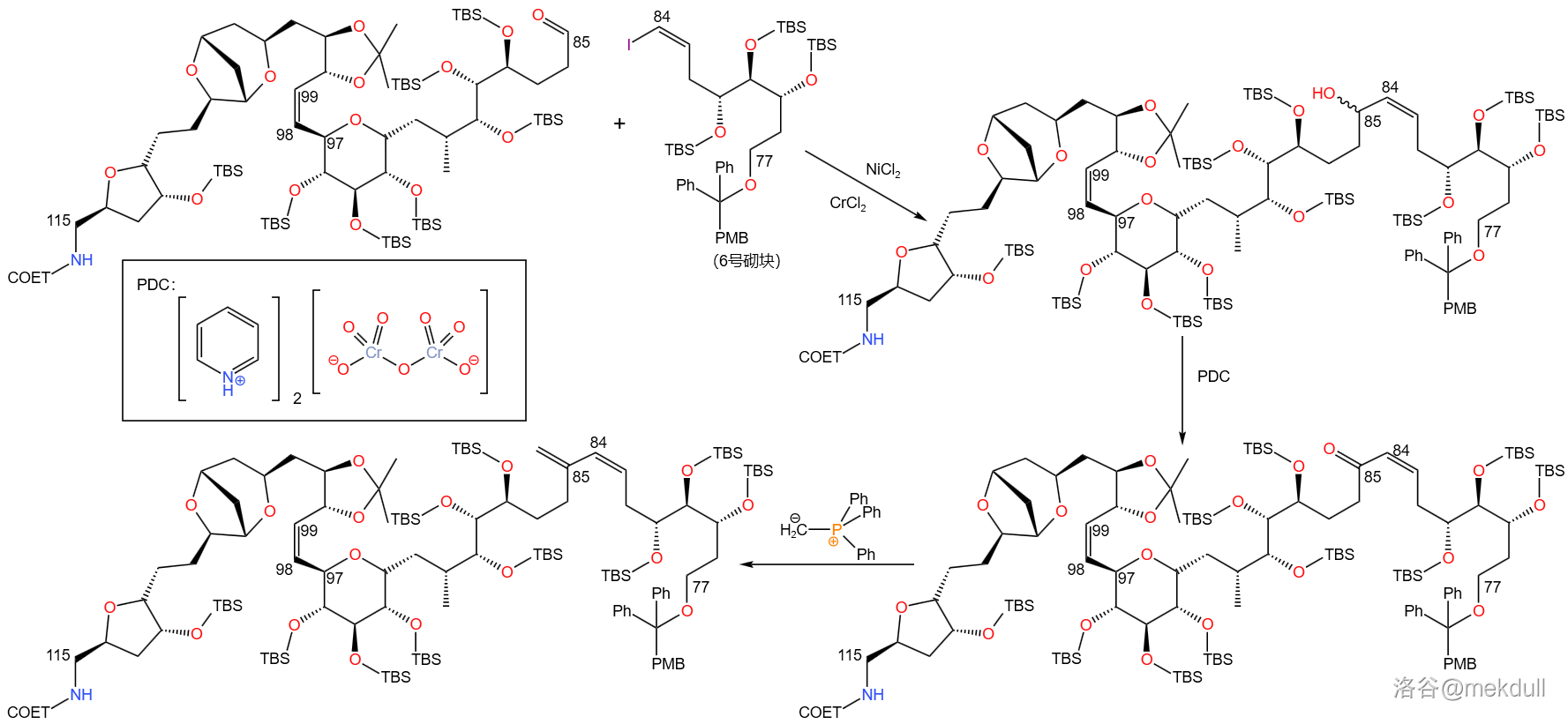
此时,我们已经成功将 $6,7,8$ 三个砌块连接到了一起。接下去便是整场全合成的真正高潮:$5$ 号砌块的连接。
观察岩沙海葵毒素的结构,不难发现 $6$ 号砌块与 $5$ 号砌块之间形成的是共轭二烯的结构,涉及 $74-77$ 号碳原子。两个双键一个顺式,一个反式。这种特殊结构怎么合成?答案呼之欲出,就是 $Suzuki$ 偶联。(中文通常翻译为铃木偶联反应)
$Suzuki$ 偶联一般是烯基硼酸或硼酸酯与烯基卤化物之间的偶联反应,如果把烯基换成芳基也可以(虽然近些年来有一些研究使得这个偶联反应可以扩展到烷基硼酸(酯)与碘化物)。$5$ 号砌块是烯基碘化物,所以另一个底物应当是烯基硼酸或硼酸酯(如下图)。那么问题就变成了,怎么把硼酸酯基团引入这个分子?

一开始,课题组考虑的是常规方法,即用末端炔烃与硼烷类物质的 $Brown$ 加成反应(中文一般成为布朗加成,是一种反马氏规则的加成反应)值得烷基硼酸酯。但当他们尝试用 $HBcat$(儿茶酚硼烷)这么做时,却发现儿茶酚硼烷总是先进攻 $115$ 号碳原子上的氨基甲酸酯基,等那边反应完了才会过来对 $77,76$ 号碳原子的末端炔烃进行加成,如下图:
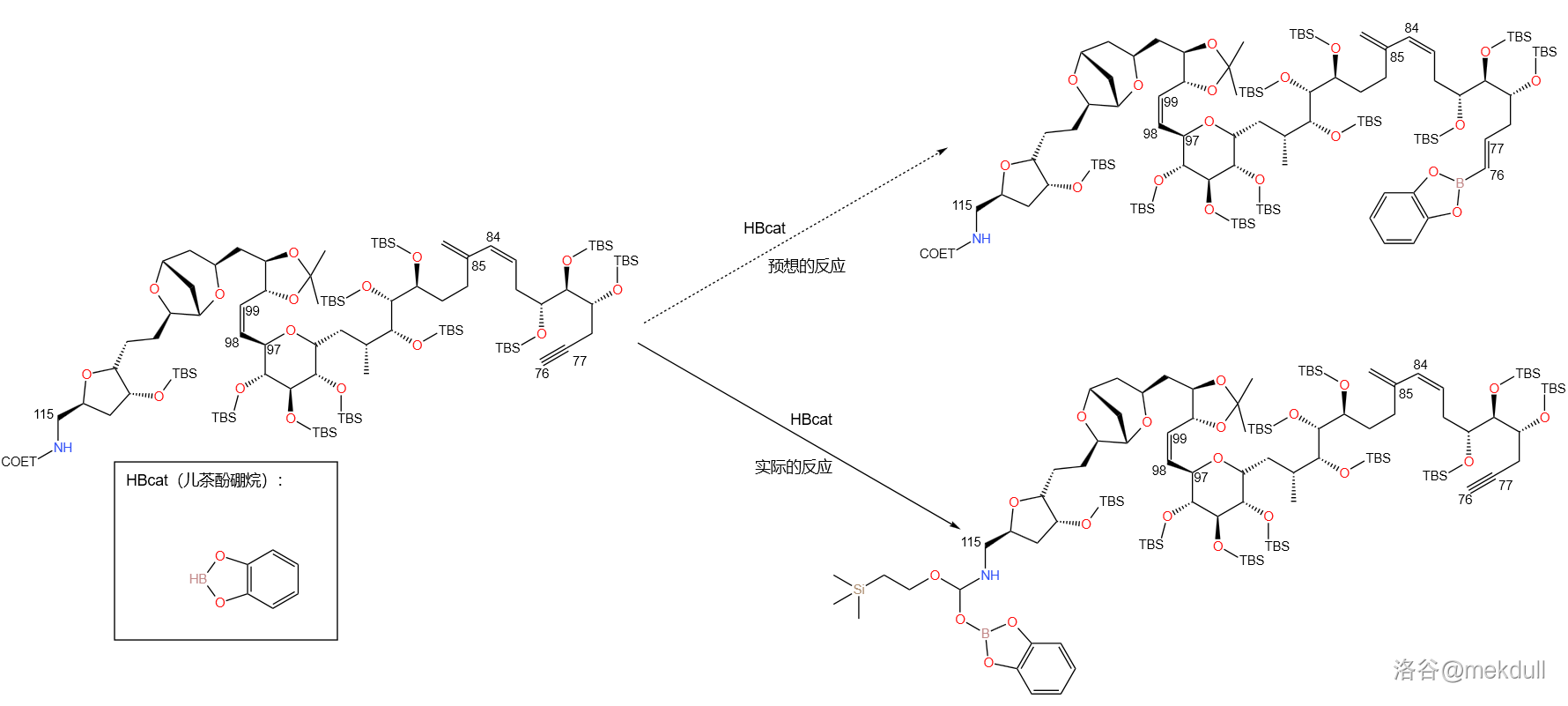
在尝试了各种反应条件、溶剂等没有效果后,课题组最后放弃了这一方法,转而使用刚出现不久的 $Matteson$ 硼酸酯化反应。这是一种从醛出发合成多一个碳原子的烯基硼酸酯的反应,若最后再使用过硼酸盐处理产物则可得到多一个同系碳原子的醛,因而也被称为 $Matteson$ 增碳反应。反应机理如下:
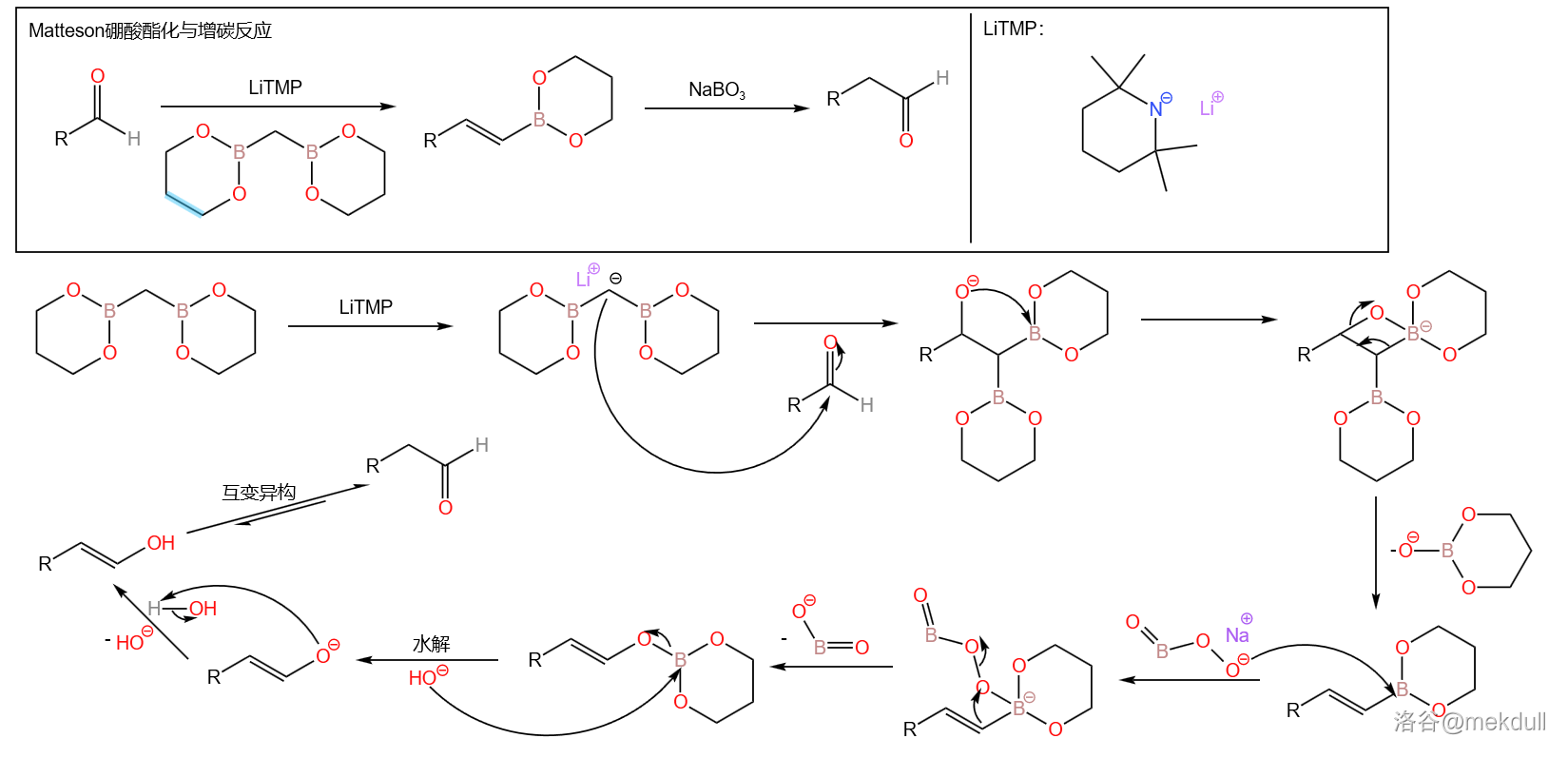
回到具体的全合成,在 $PPTS$ 的作用下,$77$ 号碳原子上的醚保护基被脱去,得到游离醇羟基,随后使用 $Swern$ 氧化在 $77$ 号位得到醛。接下来便是上面所说的 $Matteson$ 硼酸酯化反应,得到了多一个碳原子($76$ 号碳原子)的反式烯基硼酸酯。最后将硼酸酯水解,得到烯基硼酸:

有了烯基硼酸,接下来就该把两块东西连在一起了。可新的问题又接踵而至:课题组在使用朴素 $Suzuki$ 偶联时,发现期望得到的产物产率极低,几乎沦落到了杂质的地步,反而是烯基碘化物的自偶联产物占据了绝大部分,如下图所示:
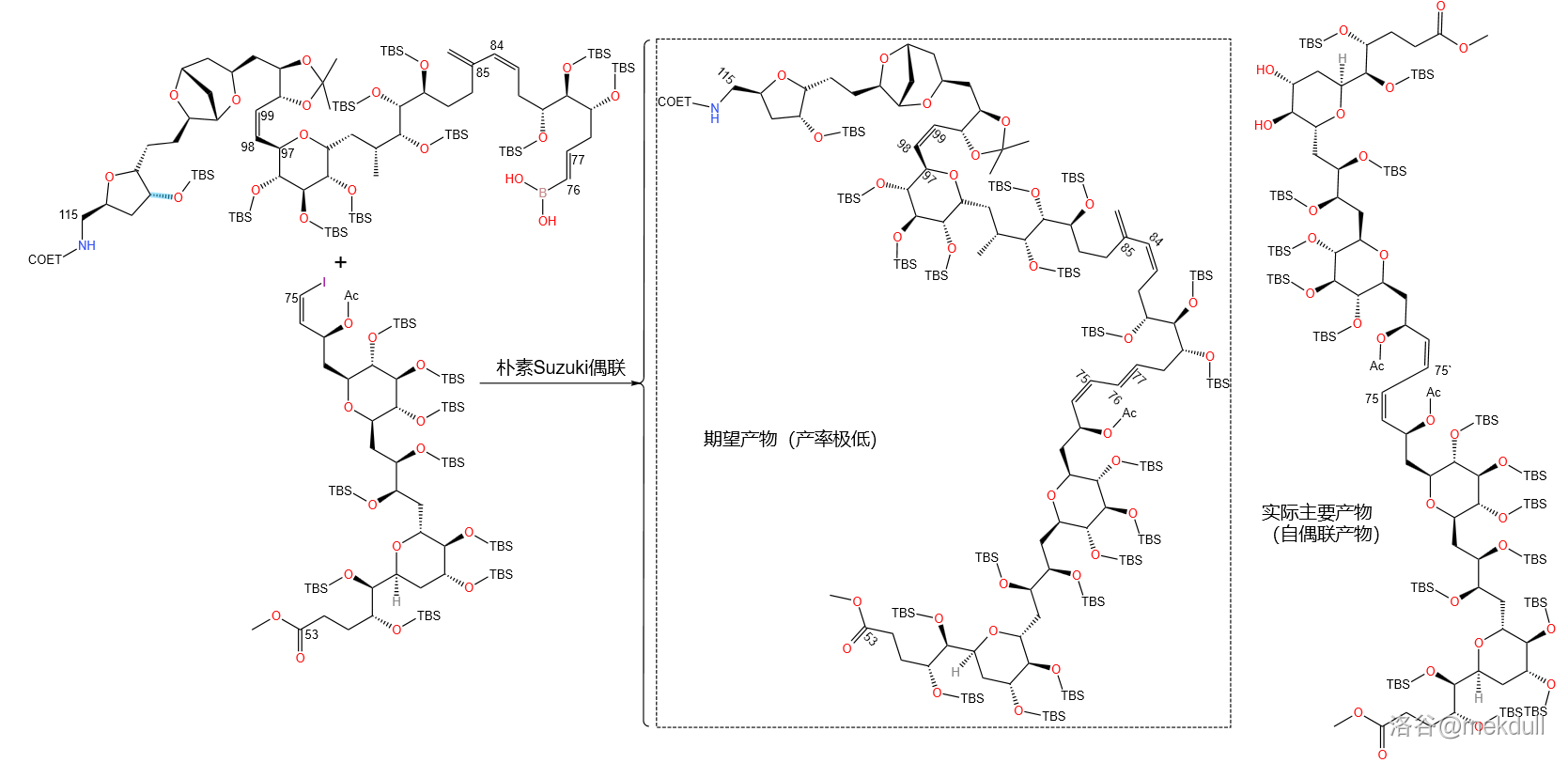
为什么会这样呢?这就涉及到 $Suzuki$ 反应的机理了。目前普遍认为的反应机理可以用一个催化环来表示,经过 $4$ 个阶段:氧化加成($oxidative~addition$)、配体交换(也有人称为配体复分解,$metathesis$)、转金属化($transmetallation$)与还原消除($reductive~elimination$)。如下图:
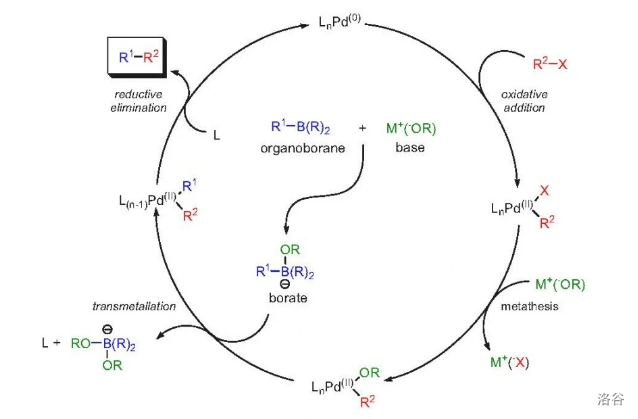
课题组推测,问题就出在前两步上。第一步氧化加成的速率较快,而第二步配体交换的速率较慢,导致了第一步得到的中间产物烯基碘化钯($\ce{X-L_{n}Pd-R}$)发生堆积。堆积之后,它便会自己与自己转金属化,随后还原消除得到自偶联产物:
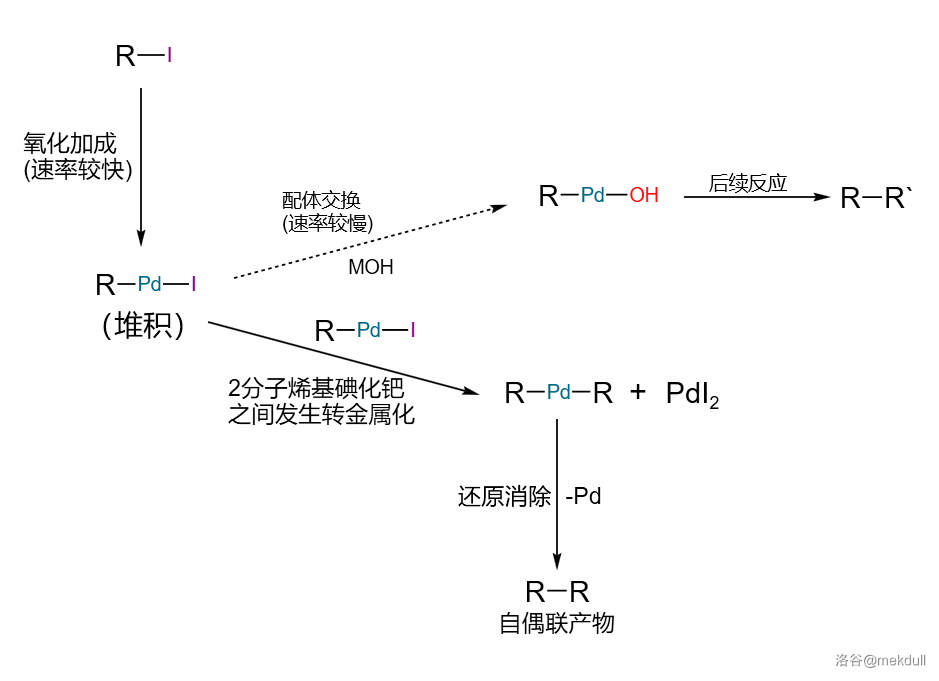
那么怎么解决这个问题呢?容易发现,我们其实只需要想方设法提高配体交换的速率,就不会发生烯基碘化钯堆积,问题也就迎刃而解了。
观察配体交换这一反应($\ce{RPdI}+\ce{MOH->MI}+\ce{RPdOH}$),我们可以发现以下三点:
-
选用不同的碱会对这个反应的速率快慢产生影响;
-
如果使用的碱如果能游离出更多的 $OH^{-}$,那么就可以提高反应速率;
-
如果使用的碱中的阳离子 $M^{+}$ 可以与碘离子牢固结合或与其形成沉淀,那么也可以为反应提速。
但这 $2,3$ 两点似乎有些矛盾:一个金属的氢氧化物如果是强碱,那么它一般就是大体积的碱金属或碱土金属了,其他金属的氢氧化物基本都是难溶性弱碱。可是,这些金属与碘的结合能力都比较差,它们的碘化物基本都可溶。事实也证明了,$\ce{KOH,CsOH,Ba(OH)2}$ 等都没有明显效果。
难道真的找不到一个有效的碱了?或者是必须要用有机碱来完成这个任务?
就在这时,课题组的眼光投向了元素周期表的另一边,并在那里找到了符合理论要求的金属离子:亚铊离子($Tl^{+}$)。
(上图:铊元素在元素周期表中的位置)
由于 $6s^2$ 惰性电子对效应,$Tl$ 虽然是硼族元素,但稳定价态却是 $+\text{I}$,这也使得亚铊离子($Tl^{+}$)有着颇为独特的性质。首先,作为软硬酸碱理论中的软酸,它与诸如 $I^{-}$ 这样的软碱结合非常紧密,$TlI$ 的难溶性即是明证;其次,作为第六周期元素,$Tl^{+}$ 半径很大,不输给碱金属离子,而它的氢氧化物也是可溶于水的强碱。而这也正好符合了理论的需要。
果然,当课题组将 $TlOH$ 当作碱加入反应体系中时,效果可谓立竿见影:自偶联产物几乎完全消失了,取而代之的即是高产率的期望产物:

如今,使用 $TlOH$ 作为碱的 $Suzuki$ 反应改进法已然被广泛用于各种复杂物质的全合成或半合成当中,这也是岸义人团队对有机合成化学做出的重大贡献。($PS:$ 包括 $TlOH$ 在内的几乎一切含 $Tl$ 的简单化合物都是剧毒甚至极毒的,如果想要尝试这种方法,请务必做好妥善的防护措施)
解决了 $Suzuki$ 偶联的问题后,$5,6,7,8$ 四个砌块就连接在一起了,整个右半部分的合成也就宣告完成了。但是,全合成的工作到此还远没有结束。接下来要做的,就是把左右两个部分给连接起来。
3.3 左右两部分的拼接
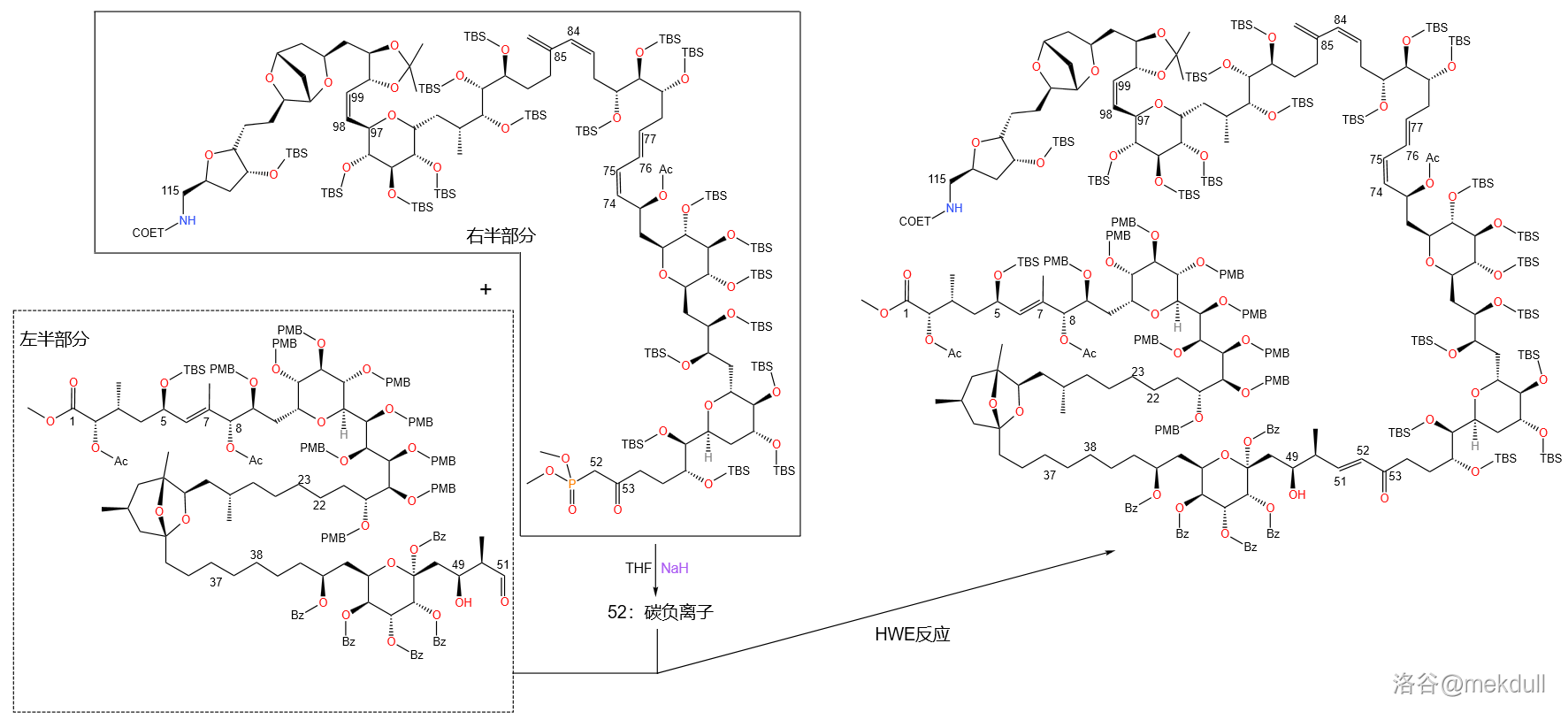
观察合成出来的两个部分,我们发现:左半部分有 $1-51$ 号碳原子,而右半部分是 $53-115$ 号碳原子,中间还缺了一个 $52$ 号碳原子。事实上,这是课题组故意那么设计的,因为接下来会使用 $Hoener-Wadsworth-Emmons$ 反应(以下简称 $HWE$ 反应)制得 $51,52$ 号位的反式烯烃。
首先,使用 $LiDMMF$(锂代甲基磷酸二甲酯)与右半部分 $53$ 号碳原子的酯基发生亲核取代,引入 $52$ 号碳原子的同时生成磷酸酯:

接着,这个磷酸酯 $52$ 号碳上的 $\alpha\text{-}H$ 在 $THF$ 中被 $NaH$ 夺去,形成磷酸酯碳负离子。这个碳负离子随后和左半部分 $51$ 号碳原子上的醛羰基发生 $HWE$ 反应,得到 $51,52$ 号上 $E$ 构型的碳碳双键:
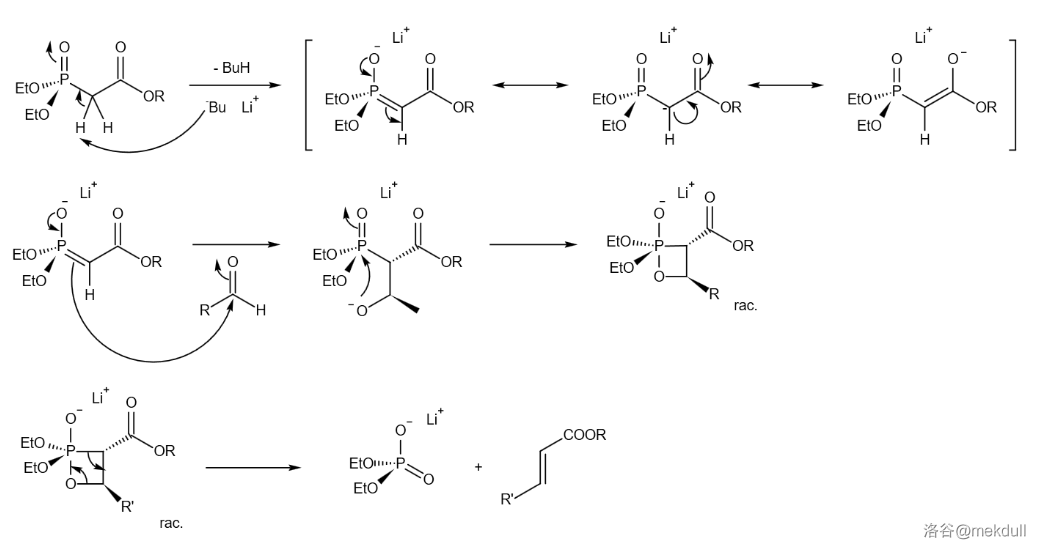
$HWE$ 反应是一种被广泛采用的变形 $Wittig$ 反应,由醛(或酮)与磷酸酯出发可以得到高产率的 $E$ 构型烯烃,且最后的产物更容易分离。反应机理如下:
$HWE$ 反应完成之后,左右两个部分 $1-8$ 号砌块就连接在一起了。最后还需要把 $53$ 号碳原子还原成羟基。课题组一开始尝试直接使用 $NaBH_{4}$ 进行还原,但 $53$ 号碳原子的手性又开始发难,最后得到的是接近 $1:1$ 的两种对映异构体。课题组还尝试使用其他烷基硼氢化钠,也没有一个可以实现不对称还原。
最后,他们还是使用了 $Luche$ 不对称还原,对稀土金属盐进行筛选。最终发现,使用三氯化铕($EuCl_{3}$)与 $LiBH_{4}$ 的组合可以有最好的选择性,比例达到了 $8:1$。还原完成以后,用 $Ac_{2}O$ 将 $53,49$ 号碳原子上的羟基保护起来:

至此,完全保护的岩沙海葵毒素羧酸的全合成宣告完成。课题组在 $1989$ 年完成了这一工作,成功也似乎已经近在眼前。

(上图:《一种完全保护的岩沙海葵毒素羧酸的全合成》论文影)
3.4 所有保护基的脱去
好,现在看看我们得到了什么?一个拥有 $7$ 种共 $42$ 个保护基的巨大分子。要把这些保护基全部脱去可不是一件容易的事,因为游离出来的羟基越多,分子就对各种试剂越敏感。
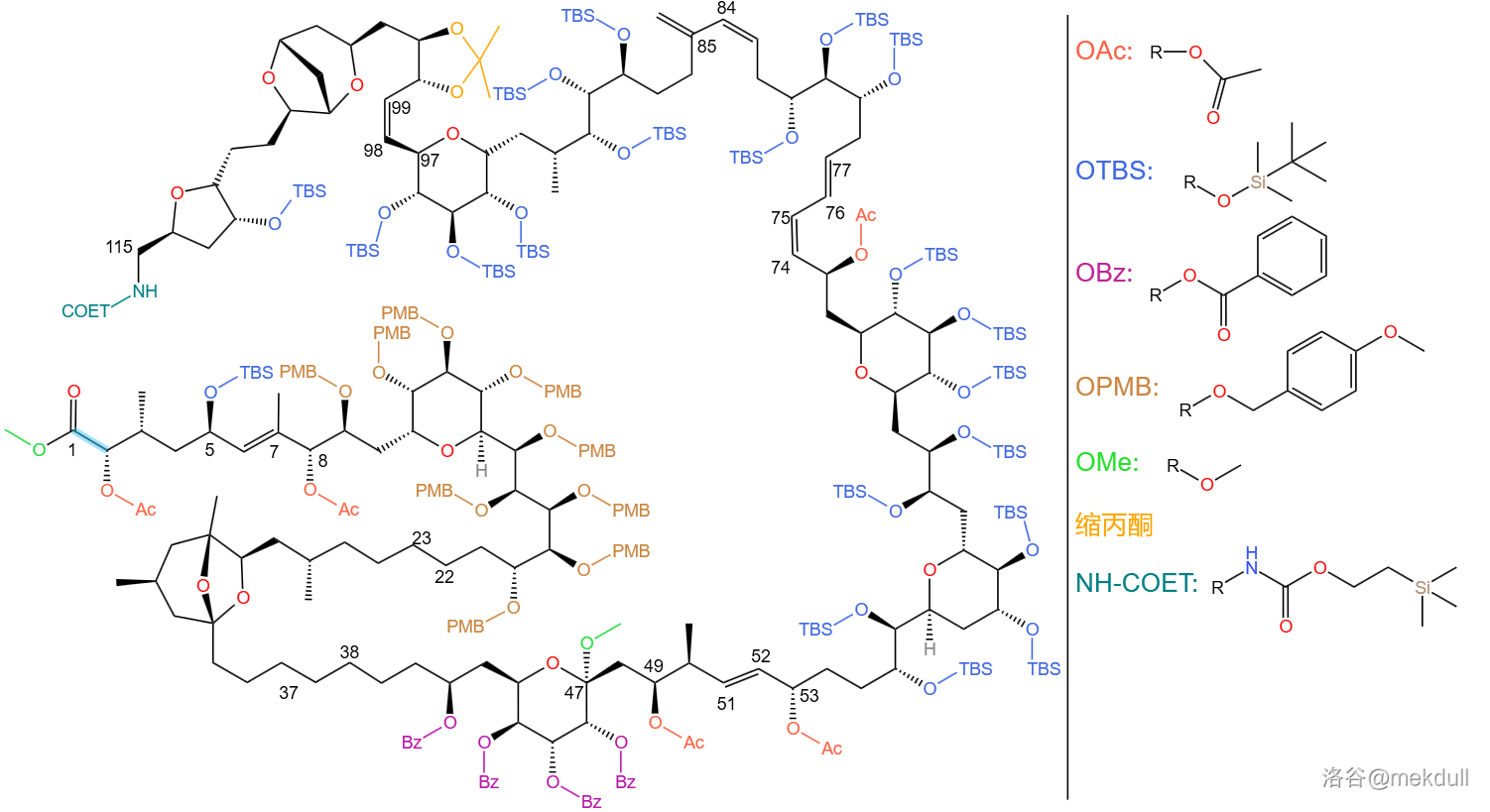
(一个错误:$47$ 号为羟基的保护基一直都是 $OMe$,但是上面有些图里画成 $OBz$ 了,在此纠正)
经过大量实验,课题组最终确定了 $5$ 个脱保护步骤:
第一步,使用 $DDQ$(二氯二氰苯醌)在常温 $CH_{2}Cl_{2}$ 与中性酸碱缓冲液中,将所有 $OPMB$ 保护基脱去:
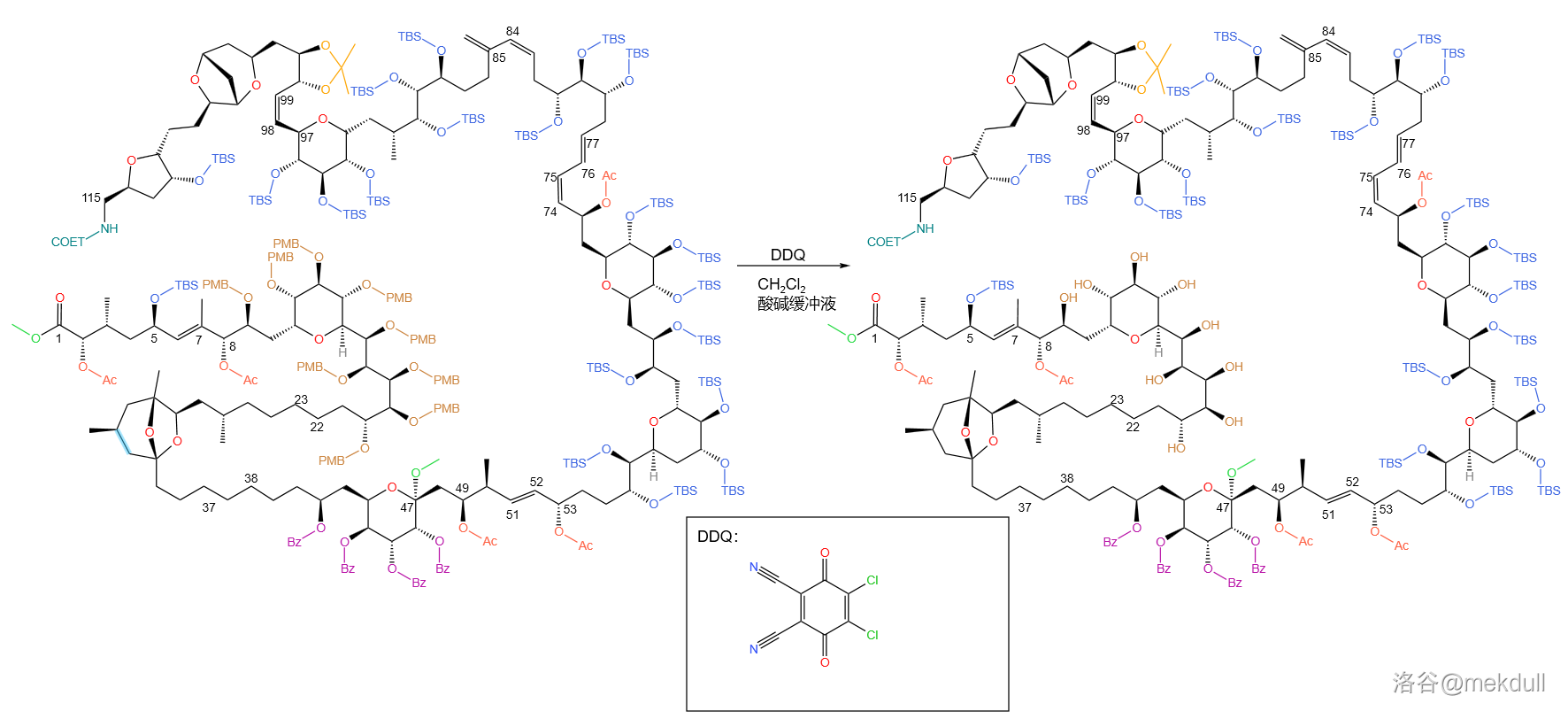
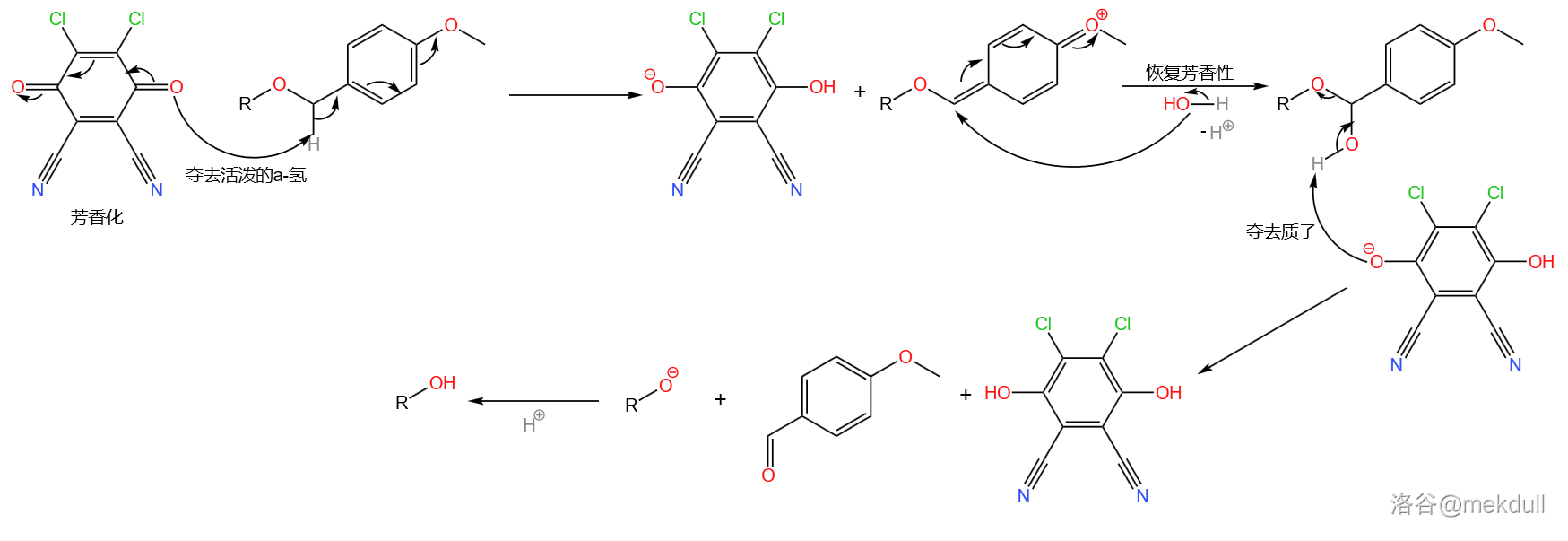
$DDQ$ 也是一个常用的氧化剂,它将 $PMB$ 保护基氧化脱去的机理可能如下:(注:最近的一些研究指出这个反应应是单电子转移($SET$)机理,本文中采用传统观点)
第二步,为了防止这些游离出来的羟基搞事,课题组紧接着又把它们用 $Ac_{2}O$ 重新保护了起来。随后,他们使用体积比 $1:12$ 的 $HClO_{4}/THF$ 水解掉了 $100-101$ 号碳原子上的缩丙酮保护:

第三步,将 $0.08mol/L-LiOH$ 溶液、甲醇($CH_{3}OH$)、乙腈($CH_{3}CN$)以 $1:2:8$ 的体积比混合,得到的混合溶液可以水解所有酯基($OBz,OAc$ 以及 $1$ 号位的甲酯):
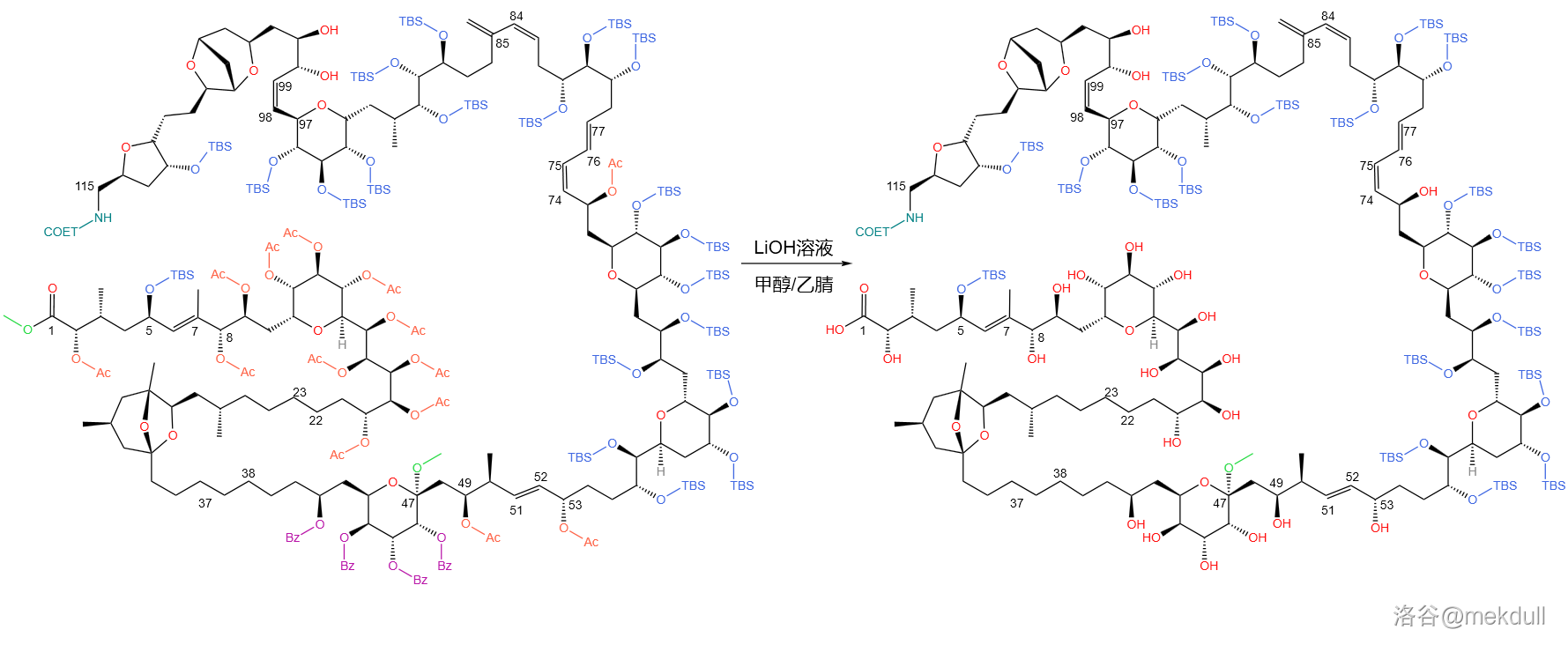
第四步,使用 $TBAF$ 的 $THF$ 溶液,脱去所有硅基保护基($OTBS$ 与 $NH-COET$):
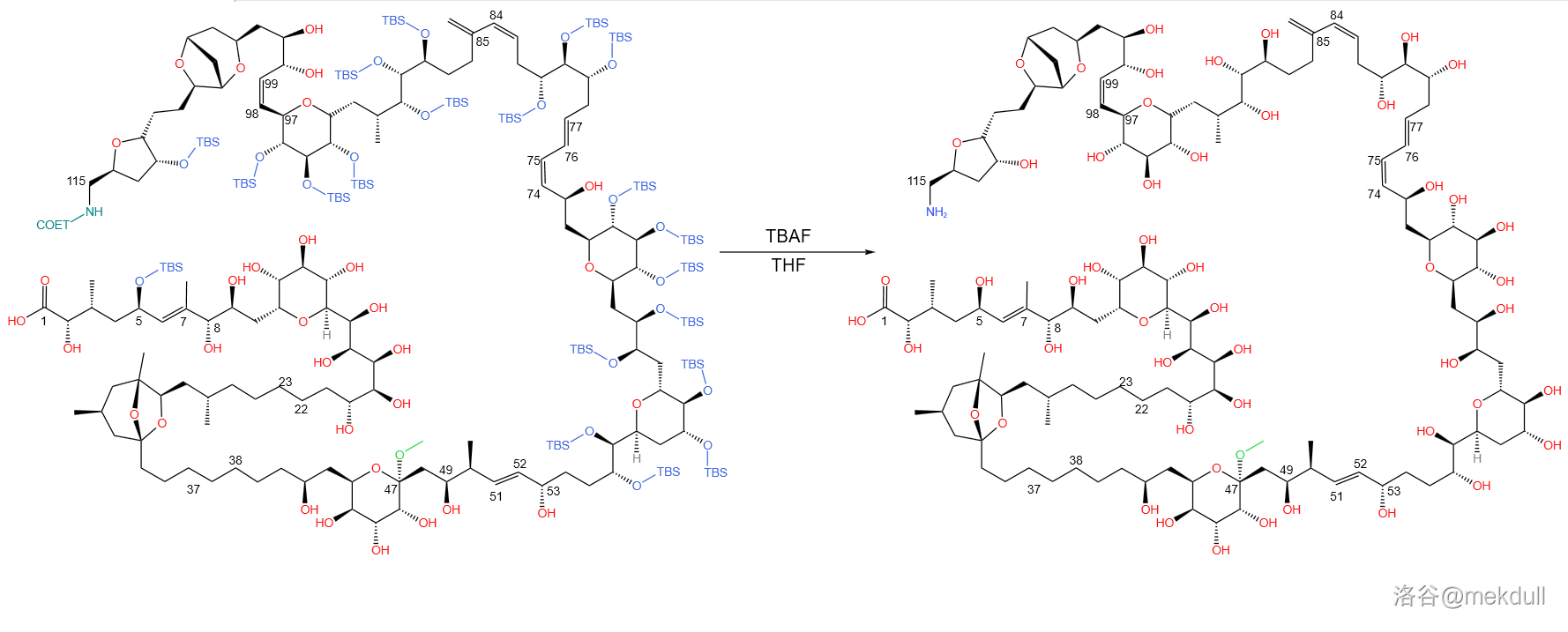
第五步,也是最后一步,使用 $1:350$ 的乙酸水溶液,水解 $47$ 号碳原子上的甲氧基:

用凝胶色谱柱除去各种盐类后,岩沙海葵毒素羧酸的全合成就大功告成了,总产率高达 $35\%$。这个工作同样在 $1989$ 年被课题组完成(事实上,两篇论文是在同一天发表的)。
(上图:《岩沙海葵毒素羧酸与岩沙海葵毒素酰胺的全合成》论文影)
现在,距离成功合成岩沙海葵毒素只有一步之遥了——连接 $9$ 号砌块。
3.5 9号砌块的连接
或许连课题组自己也没有料到,这最后的临门一脚居然花掉了他们整整 $5$ 年的光阴。作为对比,课题组从 $1982$ 年开始这项工作,在 $1989$ 年用时 $7$ 年完成了岩沙海葵毒素羧酸的全合成,即完成了 $8$ 个砌块的连接。究竟是什么困难麻烦了他们那么久呢?
观察岩沙海葵毒素的结构,可以发现 $1$ 号碳原子和 $9$ 号砌块的 $a,b$ 碳原子之间形成的是酰烯胺结构,如下图:
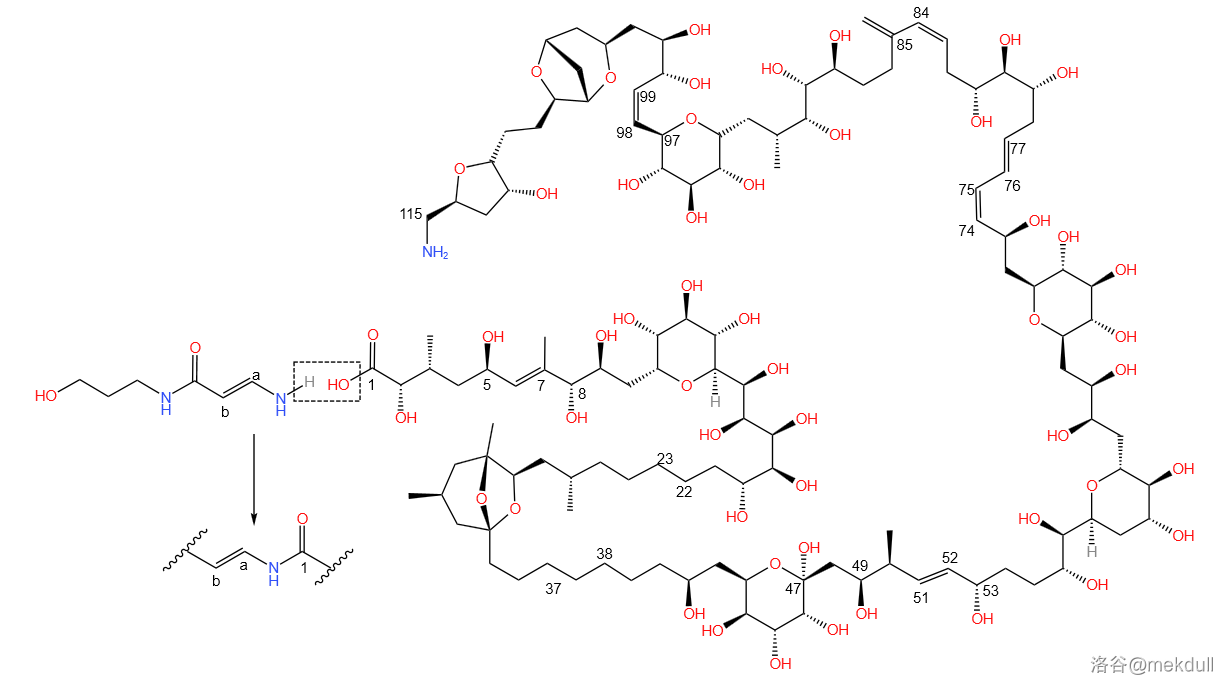
这就导致,如果要进行酰胺化反应制取岩沙海葵毒素的话,所对应的胺是一个烯胺。和烯醇一样,烯胺可以发生互变异构生成亚胺,且反应平衡偏向亚胺方向:
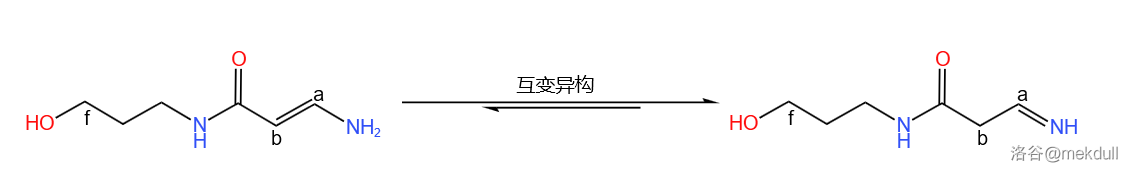
这就导致,直接使用烯胺与岩沙海葵毒素羧酸进行酰胺化反应是几乎得不到岩沙海葵毒素的。此外,由于保护基已经脱去,底物分子带有大量羟基,非常脆弱,所以反应条件必须温和。
经过大量研究,课题组确定了潜在双键(即在双键位置先连接官能团,完成酰胺化反应后将官能团消去恢复双键)的合成策略,并最后将砌块 $9$ 设计成了苯基硒化物(事实上,课题组设计了多种路线,但使用苯基硒化物产率最高)。将反应物在吡啶(溶剂)中进行漫长的酰胺化反应后,酰胺键形成,而原有的分子结构基本没有遭到破坏:
下一步当然是要将苯硒基消去。此时一般使用的是 $Clive-Reich-Sharpless$ 烯合成反应(简称 $CRS$ 反应),将苯硒基氧化消除。可这个反应中使用的氧化剂一般是过氧化物(比如 $H_{2}O_{2},m-CPBA$)或者高碘酸盐(如 $NaIO_{4}$)之类的“猛人”,对于这种羟基一堆的复杂分子来说,这样的氧化剂会把不该氧化的地方也氧化掉。在大量实验后,课题组最终放弃了这种方法,转而使用当时还比较冷门的 $Davis$ 氧化反应。这个反应使用的氧化剂樟脑磺哑嗪($Camphorylsulfonyl~oxaziridine$,又名戴维斯试剂)一般是用来温和地氧化活泼的 $\alpha-H$ 的,但同样可以氧化苯硒基并将其消除:

氧化消去的机理可能如下:

现在,酰烯胺结构被我们合成出来了,但还没有结束。这个反应生成的碳碳双键顺反选择性并不好,得到的产物中顺反比约 $2:3$。为了提高反式比,课题组采用了光异构的方法,装置如下图所示:

(注:这个装置是简化过后的,实际会更加复杂;氯化亚锡溶液是用来滤光的)
将这个装置在 $37^{\circ}C$ 下用波长为 $300nm$ 的紫外线长时间照射,所需的反式异构体比例提高,反式比顺式达到了 $6:1$。到这里,岩沙海葵毒素的全合成才终于宣告完成。这一年是 $1994$ 年。
4.总结与后记
岩沙海葵毒素的全合成是有机合成化学发展中的一块里程碑。它拓展了有机合成中分子大小、复杂度的前沿,同时给合成方法与构象分析等领域带来了重大发展。岩沙海葵毒素的全合成是一个足以载入史册的成果。
或许有很多人会问,将这些东西合成出来究竟有什么意义?能给我们的生活带来什么好处吗?在我看来,完成了一种物质的全合成就相当于拥有了它的力量,即使我们还不知道这种力量应该如何恰当地释放而服务于人。想象一下,若是哪天沙群海葵灭绝了,此时如果我们还没有完成这项工作,那么存在于这种物质中的力量或许就会被永久尘封。在全合成的基础上,我们以岩沙海葵毒素为基础,可以将其改性,从而取其精华,去其糟粕。无数良药就是这样被人类发明。关键在于,全合成的完成让人类从此有了对这种物质的自主产权。
当然,除了岩沙海葵毒素,岸义人教授的团队还在许多天然产物的全合成上做出了卓越的贡献。比如河鲀毒素(1972)、软海绵素$B$(1992)等等:(他们好像真的很执着于天然剧毒物质)

目前,以软海绵素$B$衍生物为主要成分的抗癌药 $Eribulin$(商品名 $Halaven$)已经上市,成为人类对抗癌症的又一把利器。
岸义人教授于 $2023$ 年 $1$ 月逝世。在去世前,他仍然执着于全合成研究,并在 $2019$ 年简化了软海绵素$B$的合成路线从原本的 $47$ 步下降至 $37$ 步。($2021$ 年,$K.~C.~Nicolaou$ 教授团队有简化到了 $25$ 步)让我们用这篇文章对他,以及所有从事全合成工作的化学家,表示崇高的敬意!