1.前情提要
河豚毒素($tetrodotoxin$,简称 $TTX$),是一种最早发现于鲀鱼(动物界,脊索动物门,硬骨鱼纲,辐鳍鱼亚纲,鲀形目,鲀科,东方鲀属)体内的生物碱,也是自然界中所发现的毒性最大的神经毒素之一(注射致死量约 $8μg/kg$)。由于鲀鱼俗称为河豚,故而得名河豚毒素。
下图表示了这种物质的结构:
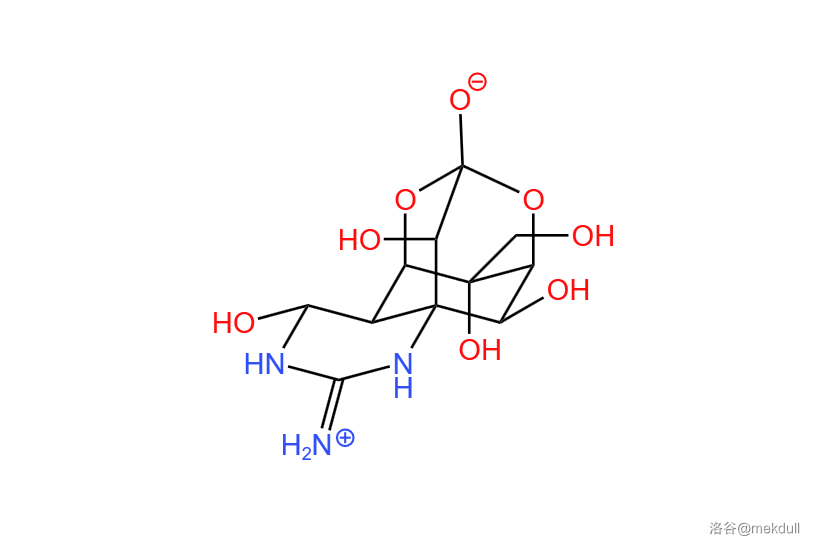
在公元前 $2500$ 年前的中国和埃及,人们就已知道有些鲀形目鱼类(河豚)是有毒的。在河豚的肝脏、精巢、血液、肾甚至是有些种类的肌肉中都含有这种物质,而织纹螺、纽虫、蝾螈等动物体内同样也有它的身影。总的来说,在漫长的演化中,许多物种都选择了用这种物质来自卫,其研究价值不言而喻。

(上图:现实中的河豚。即使身怀剧毒,也依然在几千年间一直是人类餐桌上的佳肴。)
河豚毒素是一种钠离子通道阻塞剂,可高选择性地阻断神经兴奋膜上钠离子通道,阻碍神经传导,从而引起神经麻痹进而致死。同时,它也具有巨大的医用价值,甚至有证据证明其可以抑制肿瘤。因此自 $1911$ 年日本化学家 $Tahara$ 提取出了这种物质之后,人们便对它开展了大规模的研究。$1965$ 年,$Goto$ 等人得到了这种物质的结构。下图是计算机渲染得到的河豚毒素 $3D$ 结构:
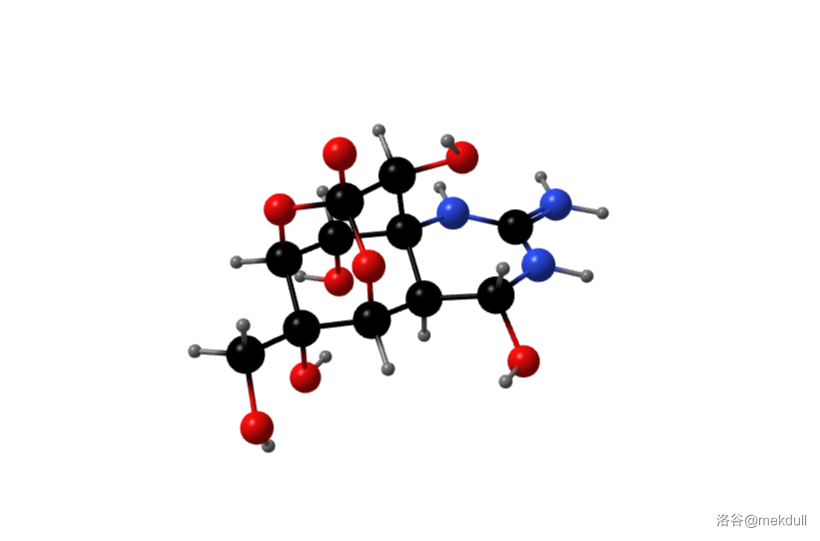
可以看见,这种物质实际以内盐的形式存在,具有笼状结构,且几乎每一个碳原子都被官能团化。这种特殊的结构赋予了其较高的化学稳定性,必须在 $200^{\circ}C$ 以上加热 $20min$ 以上才能全部将其破坏。但同时,这种物质对碱极不稳定,碱性条件下可将其快速分解,而这又给全合成工作带来了挑战。
但是,人类不会因为困难而放弃。
$1972$ 年,来自哈佛大学的日裔化学家岸义人($Y.Kishi$)带领的课题组首次成功合成了外消旋的河豚毒素。$31$ 年后,$2003$ 年,$J.Du~Bois$ 教授带领课题组完成了河豚毒素的不对称合成。在下文中,我们将重点讨论 $Kishi$ 教授的外消旋河豚毒素全合成路线。
2.概述
面对这种复杂的物质,$Kishi$ 教授选择了逆合成分析的方法。所谓逆合成分析,就是将分子“切”成几个部分,确定所需的合成碎片,然后再把它们拼起来得到目标产物。

(上图:河豚毒素的逆合成分析,那条虚线就是切开分子的“刀”)
观察结构,可以发现河豚毒素是一个高度氧化的物质,对应的合成手段便是环氧化与环氧化合物开环。在合成中,这种合成策略被多次使用。
全合成中涉及的主要反应清单:
-
$Diels-Alder$ 反应;
-
$Beckmann$ 重排反应;
-
$\ce{SeO2}$ 对甲基的氧化反应;
-
$m-CPBA$ 对碳碳双键的环氧化;
-
环氧化合物的开环;
-
$Baeyer-Villiger$ 氧化;
-
$Meerwein$ 烷基化($Meerwin’s~salt$);
接下来,就让我们正式进入全合成的介绍环节。为了方便讲解,我们会把整个全合成过程分为 $4$ 个部分。
3.逐步合成分析
3.1 第一部分:三环骨架的合成
合成的起始物是一种对苯二酚的衍生物。这种物质首先发生羟胺化反应生成肟,随后酚被 $\ce{Ag2O}$ 氧化为醌。两步反应产率约 $79\%$:
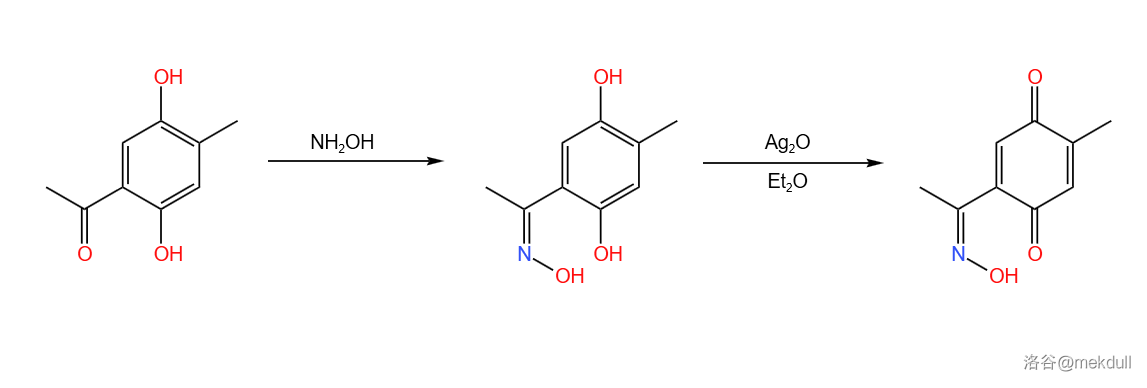
接下来,课题组打算用 $Diels-Alder$ 反应给这个物质增一个环。但此刻出现了一个问题:这个物质有两个双键都可以成为亲双烯体,而如果直接反应的话,由于肟基是吸电子基团,反应会优先在右边的双键上发生。于是,课题组加入了 $Lewis$ 酸 $\ce{SnCl2}$ 与肟基配位。这样一来,肟基的吸电子能力减弱,反应可以在左边双键上高选择性地进行:
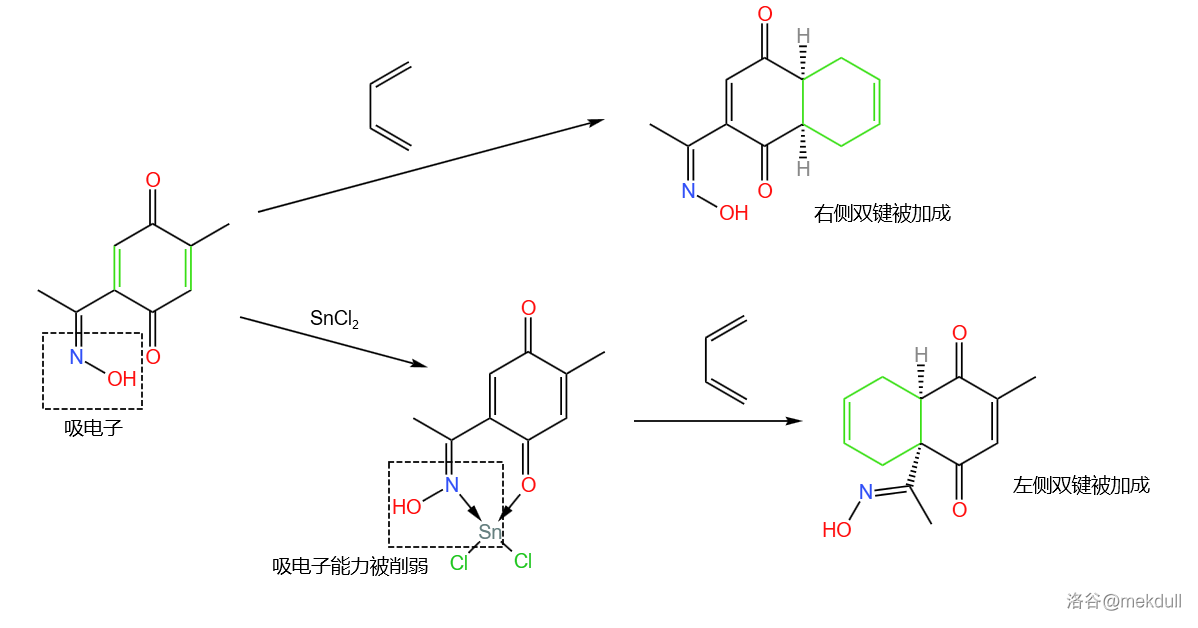
该步反应产率 $83\%$。
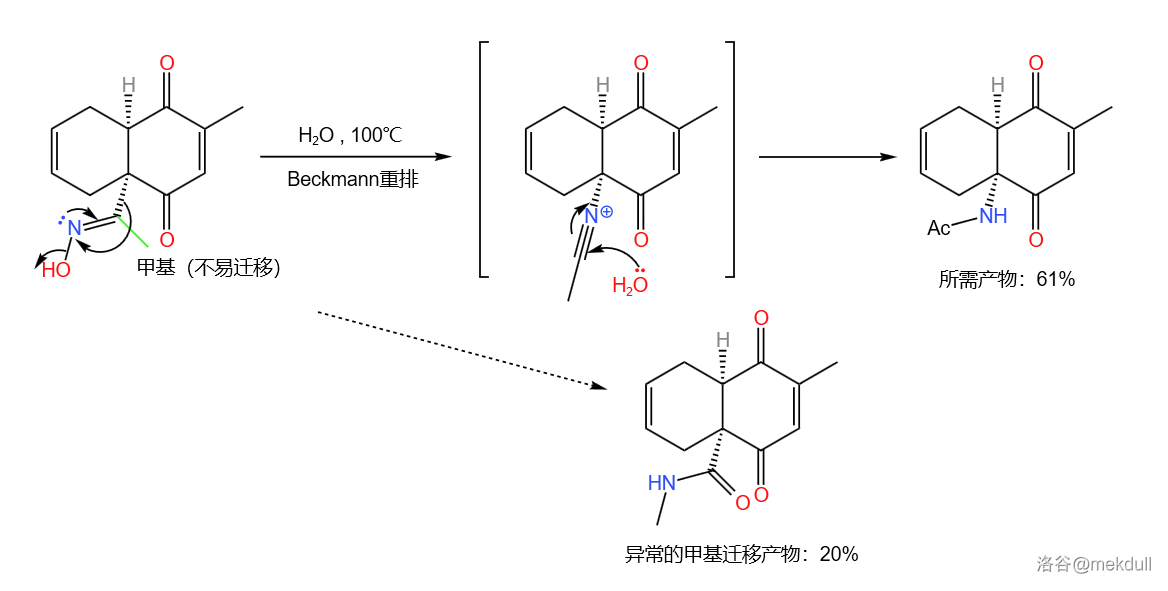
下一步,使用的是著名的 $Beckmann$ 重排反应。由于是在酸性条件进行,肟的 $E/Z$ 转换非常快,因此反应会发生在相对容易迁移的取代基上。由于底物的其中一个取代基——甲基是非常不容易迁移的基团,所以可以以较高产率得到所需产物:
随后,在 $0^{\circ}C$ 左右,用 $\ce{NaBH4}$ 可以选择性地还原其中一个羰基,以 $96\%$ 的产率得到醇。这可能是因为另一个羰基的 $\alpha-C$ 原子上连接了一个大基团,导致其空间位阻大而难以进攻:

下一步,课题组使用 $m-CPBA$(一种过氧酸)将分子左侧的双键环氧化(右侧双键可能是因为其上有取代基而拥有较大的空间位阻,所以未被环氧化)。环氧化后,在酸性条件下,分子内会直接发生环化与环氧化合物开环的反应,从而构建起一个五元环:
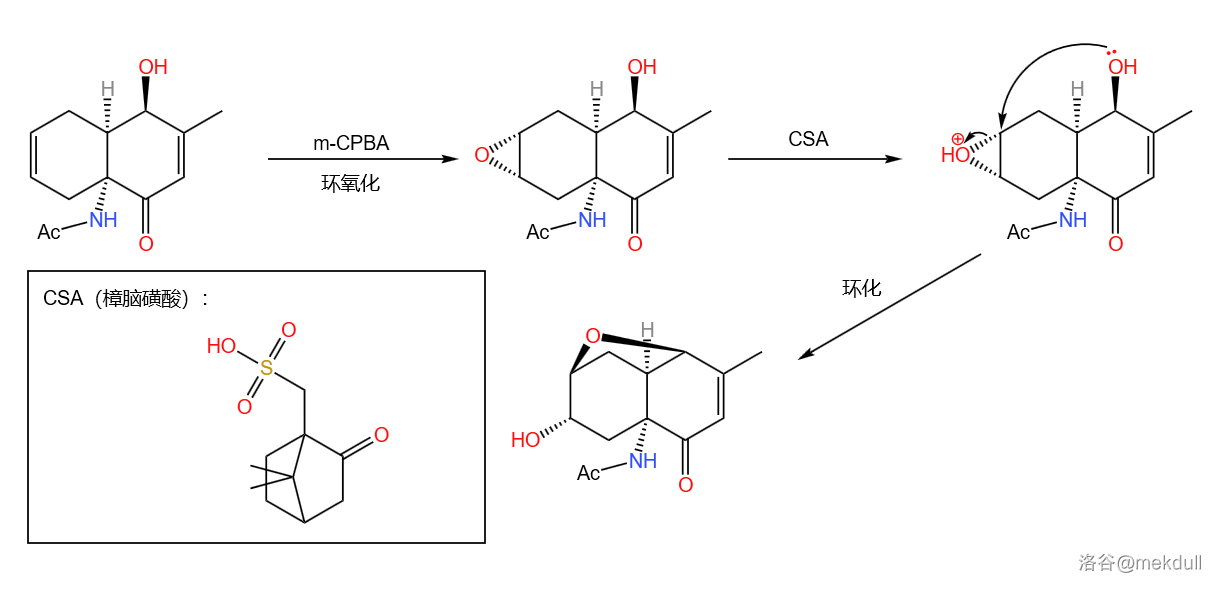
到这一步,目标分子的三环骨架已经成功建构,对应的是逆合成分析中的中间体 $107$。
3.2 第二部分:三环骨架的修补
这一阶段的任务就是对刚才得到的三环骨架进行一些缝缝补补,从而为下面的合成做铺垫,算得上是一个承上启下的部分。
首先,用 $Sarret$ 试剂将醇氧化,并用乙二醇将氧化出来的羰基保护起来。两步反应产率 $90\%$:
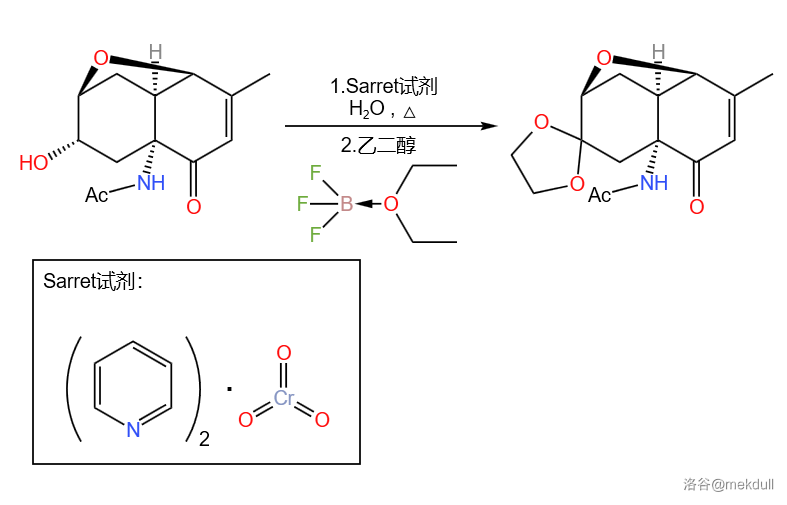
紧接着这步氧化的是 $MPV$($\text{Meerwein-Ponndorf-Verley}$)还原,把另一个 $\alpha,\beta-$不饱和的羰基用异丙醇铝($\ce{Al(O}i\text{-}\ce{Pr)3}$)、异丙醇($i\text{-}\ce{PrOH}$)处理为羟基,再将它乙酰化保护:
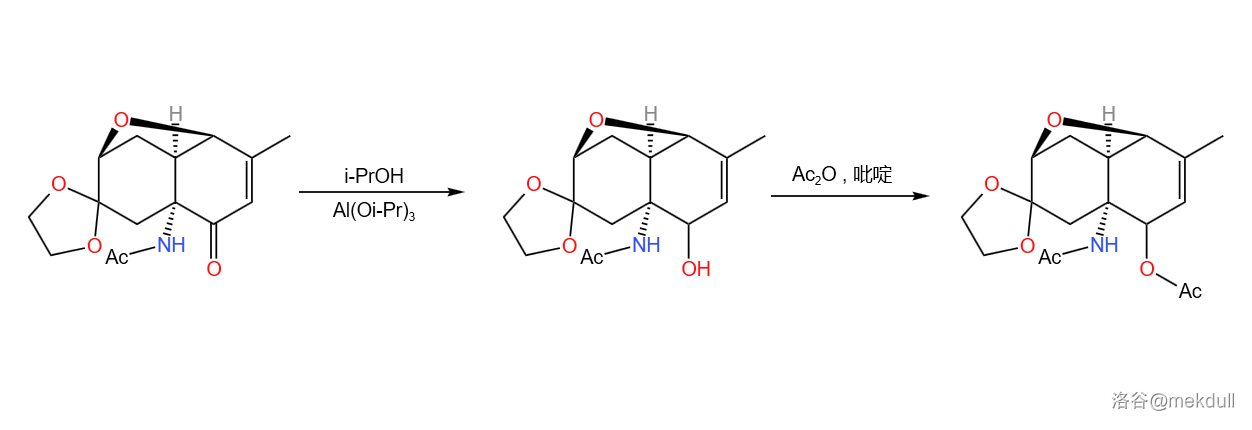
上步反应产率约 $95\%$。$MPV$ 反应的机理如下。由于反应实际上是可逆的,所以课题组加入了过量的异丙醇使反应得以正常进行。

随后,使用 $\ce{SeO2}$ 对分子双键上的甲基进行氧化,得到 $\alpha,\beta-$不饱和醛;醛又在 $\ce{NaBH4}$ 作用下还原,得到 $\alpha,\beta-$不饱和醇:
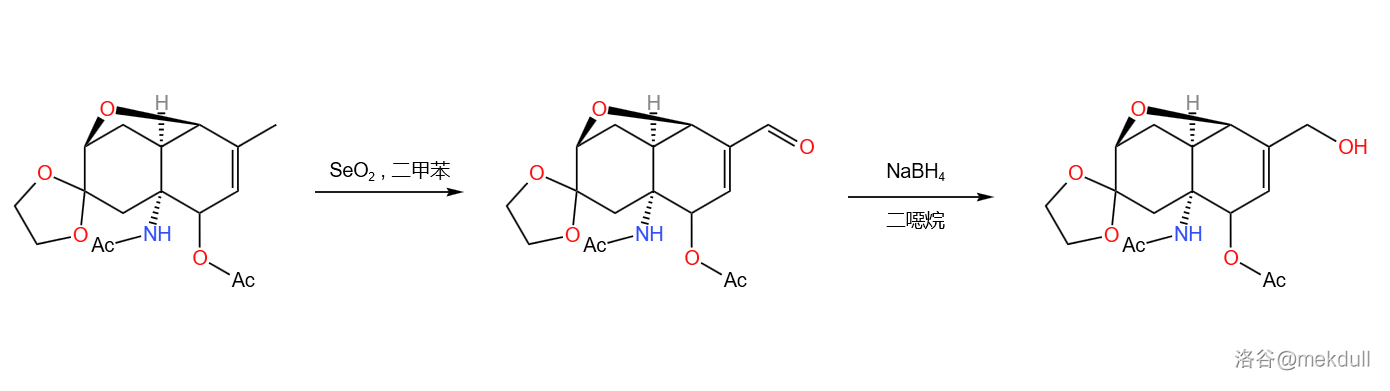
下一步,是将分子中的双键环氧化。可问题是,这个双键可能是因为空间位阻的原因,具有很高的惰性,需要加热;但一加热,就会导致 $m-CPBA$ 的分解,进而导致产率下跌。于是,$Kishi$ 课题组筛选了许多自由基抑制剂,试图阻止 $m-CPBA$ 的分解。
最后,课题组发现一种工业上被称为抗氧剂300(化学名硫代双特丁基间甲酚)的物质效果最好,它使得反应可以在 $90^{\circ}C$ 条件下进行,产率高达 $95\%$:

环氧化之后,是一系列保护——脱保护——换保护的步骤。将羟基乙酰化;脱去缩酮保护;将暴露的酮羰基再用乙醇缩合保护:

为什么要换保护?是为了这一步在 $180^{\circ}C$ 的邻二氯苯($\ce{C6H4Cl2}$)中,分子发生消去反应,消去 $1$ 分子 $\ce{EtOH}$ 产生双键:
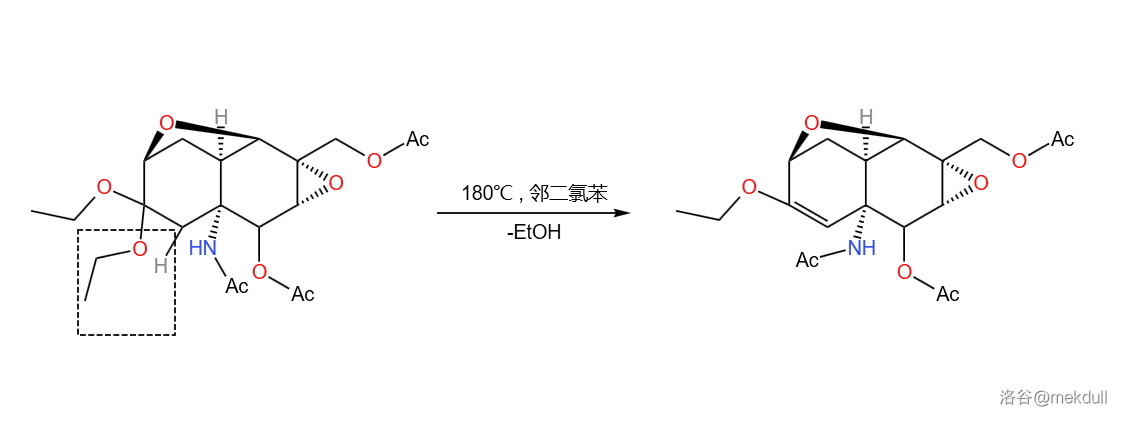
两步反应总产率 $80\%$。
至此,第二阶段的合成也宣告完成。在下一个阶段中,我们就将完成河豚毒素基本骨架的搭建。
3.3 第三部分:完成河豚毒素骨架的搭建
上一阶段的最终产物在 $m-CPBA$ 与 $\ce{AcOH}$ 的作用下,酮羰基被还原,并在 $\alpha$ 位导入一个乙酰化保护的羟基:
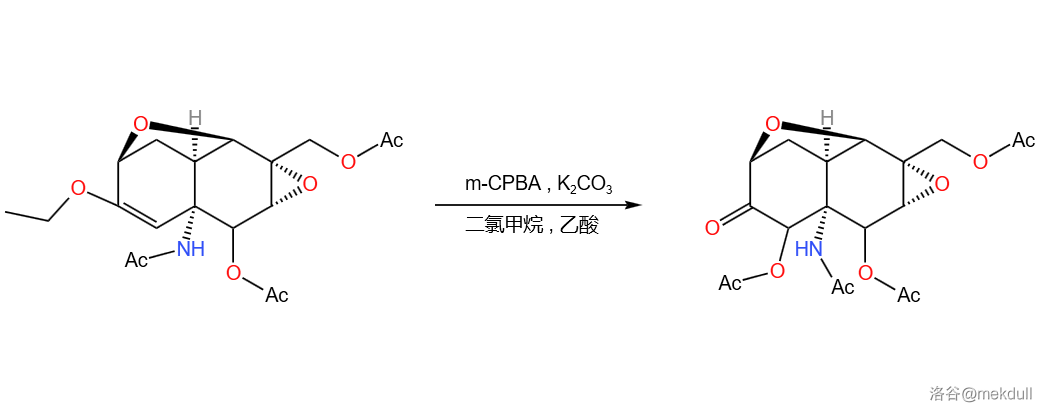
反应的机理如下,经历环氧化物中间体:

紧接着,使用 $m-CPBA$ 对该物质进行 $Baeyer-Villiger$ 氧化,将六元环扩大为七元环内酯:
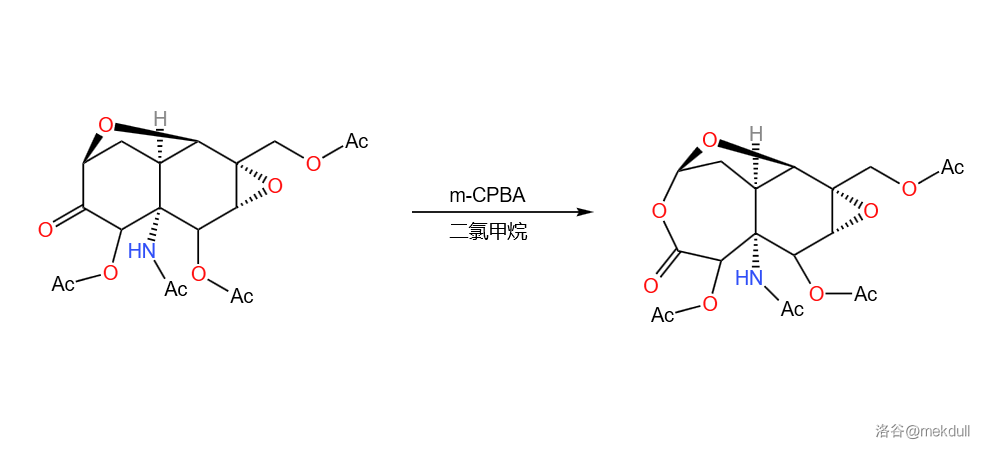
$Baeyer-Villiger$ 氧化的机理如下:(论文链接:J. Am. Chem. Soc. 1972, 94, 12, 4189–4196)

随后,在乙酸钾($\ce{KOAc}$)与乙酸的作用下,内酯断裂,生成的氧负离子随即进攻另一侧的环氧基使其开环,完成桥的构建:

接着,用 $\ce{Ac2O}$ 乙酰化保护羟基,第三部分的合成就完成了:
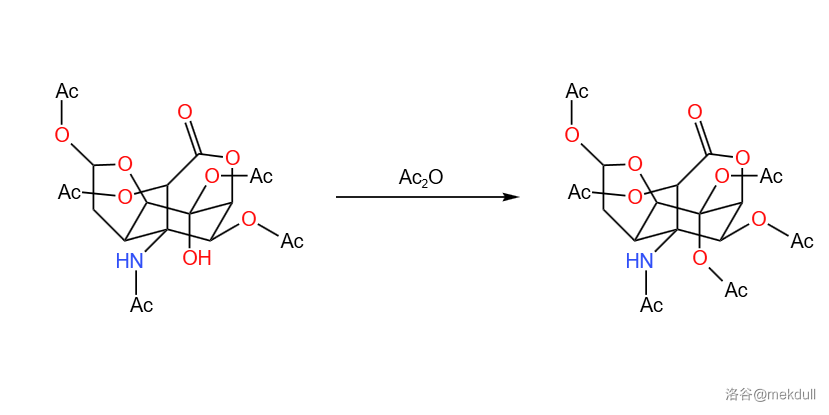
最后,在 $300^{\circ}C$ 的温度下,分子发生消去反应,恢复左侧五元环醚中 $\alpha,\beta$ 位的双键。虽然在分子中有很多 $AcO-$ 基团,但观察结构即可发现左侧的这个基团是最容易消去的:
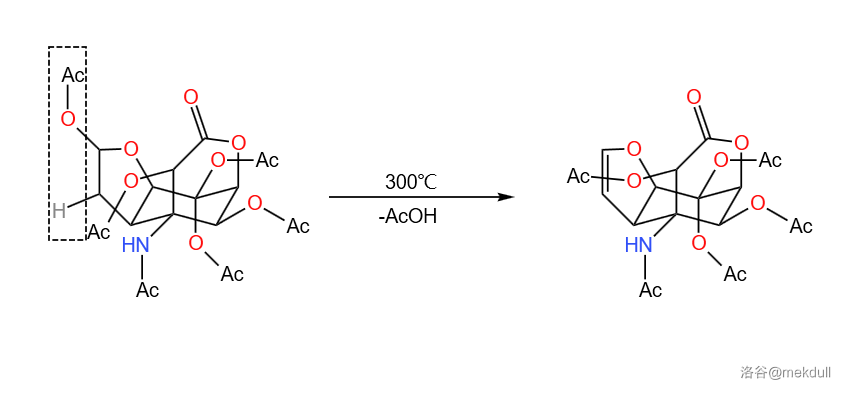
这一部分的五步反应总产率 $56\%$。自此,我们完成了河豚毒素碳骨架的构建。
3.4 第四部分:完成河豚毒素的全合成
首先,上阶段合成的最终产物经过梅尔文盐($Meerwein’s~salt$)处理,$N$ 原子上连接的乙酰基被烷基化。随后让其在酸性条件下水解,两步反应效果相当于脱去了 $N$ 原子上的保护基:
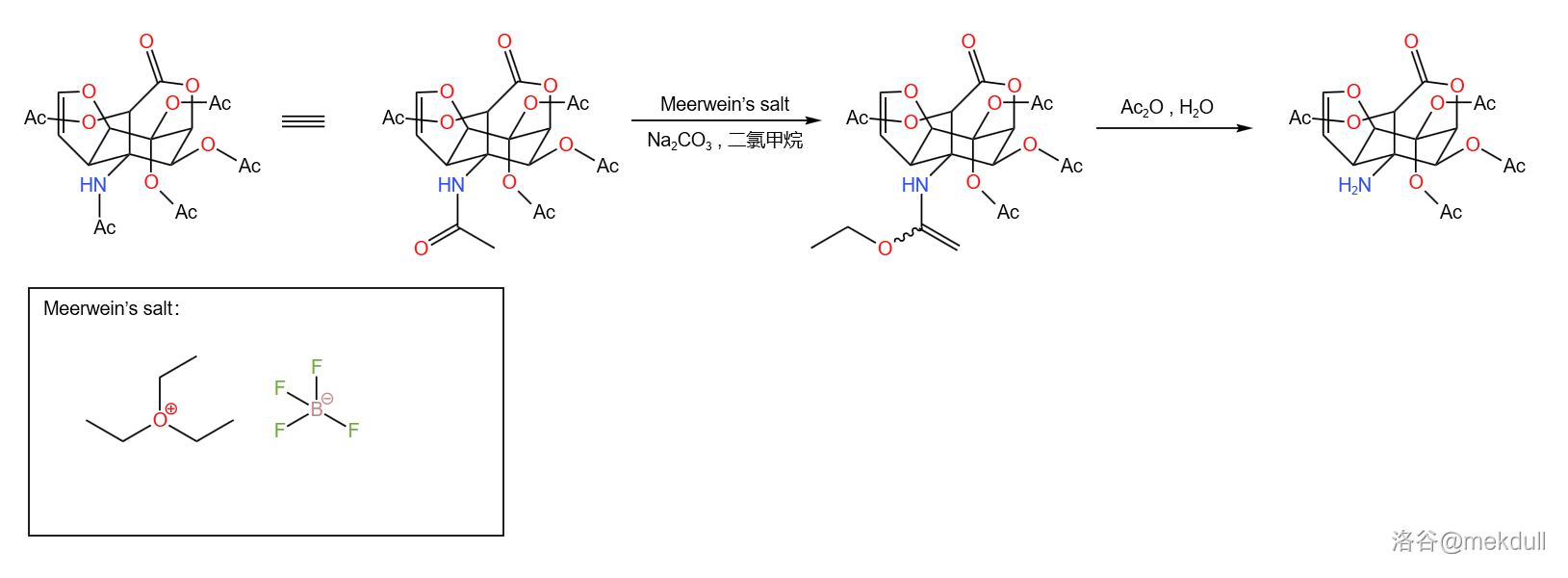
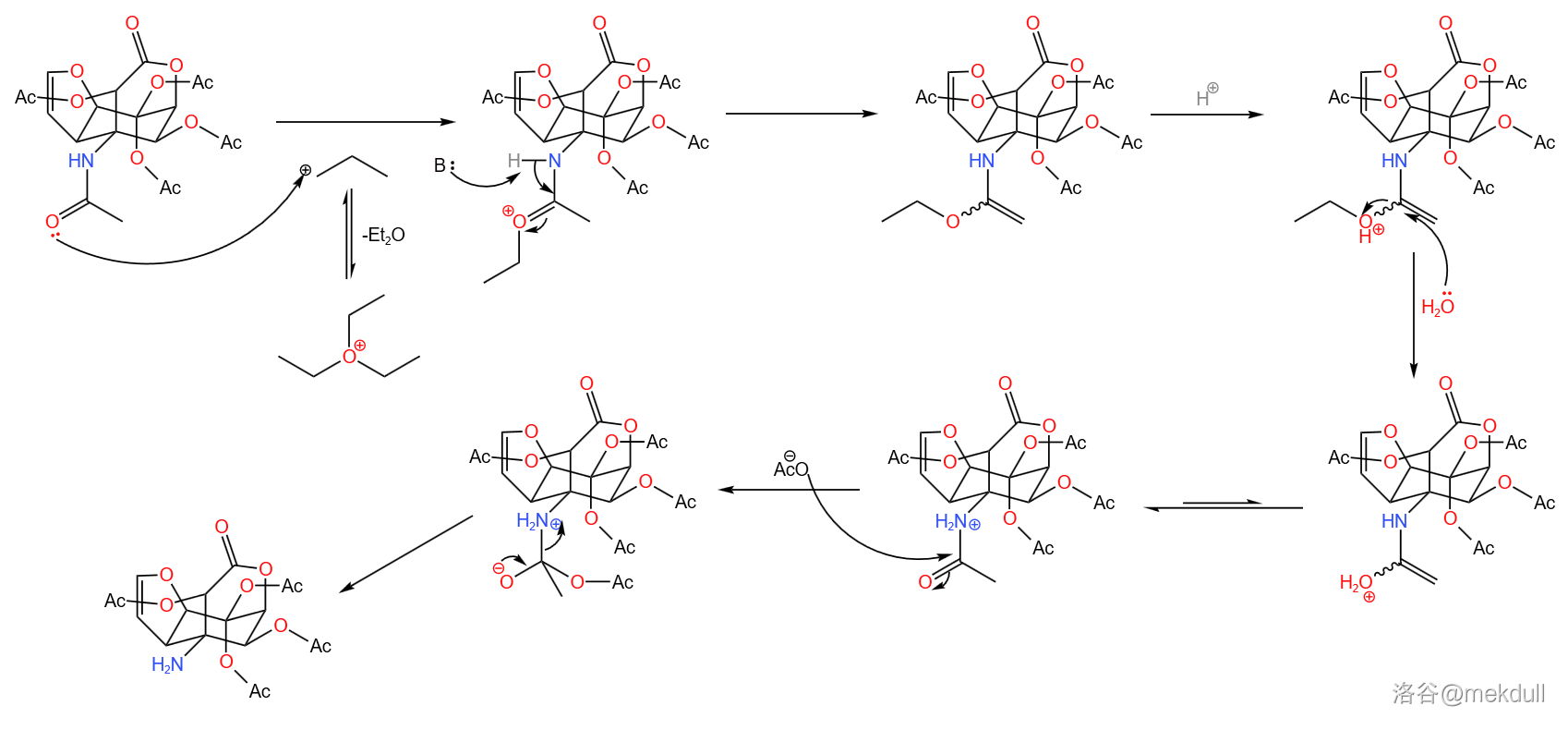
反应机理如下,总产率 $92\%$:
随后,该分子与一亚胺化合物在 $120^{\circ}C$ 下反应,接着再与乙酰胺($\ce{AcNH2}$)反应得到胍基化合物。反应与其机理如下:
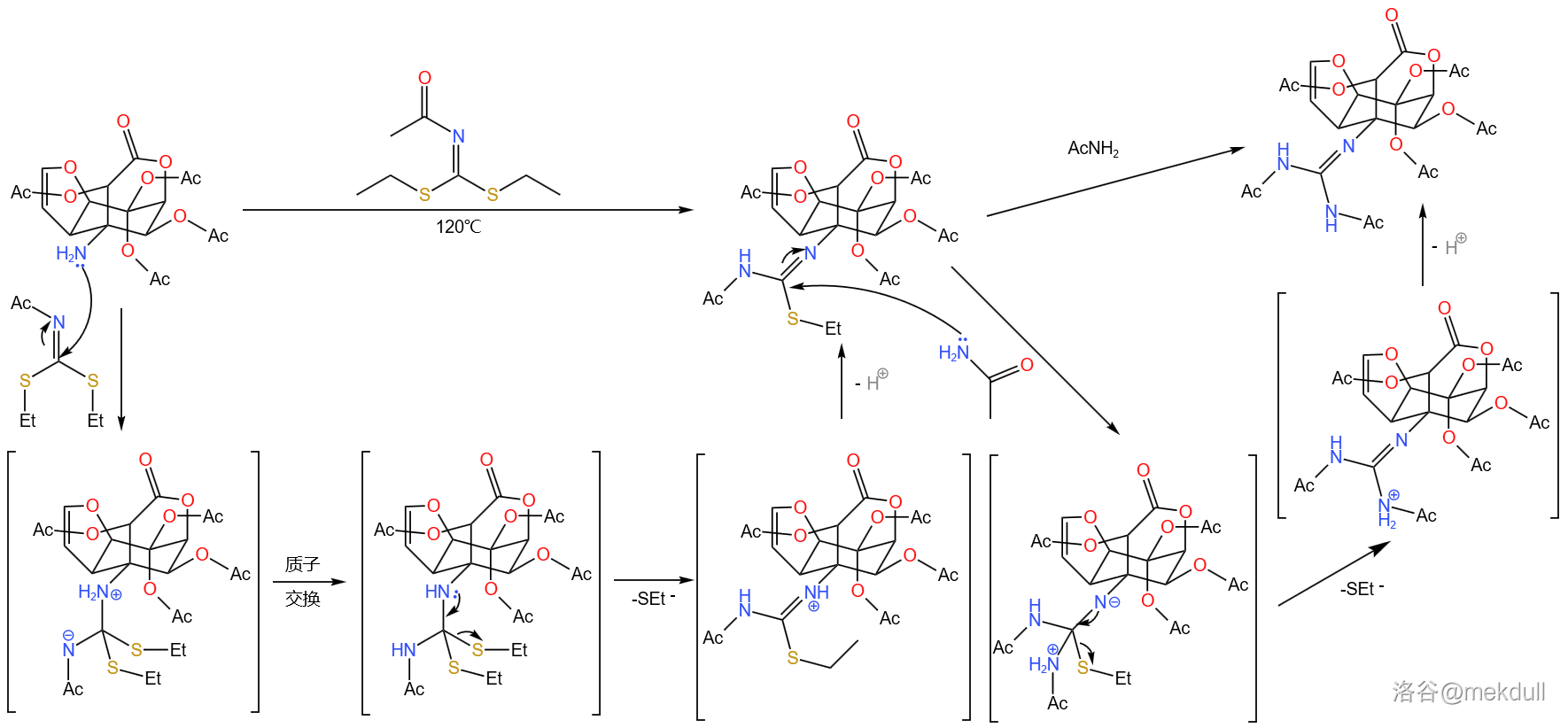
两步反应产率 $18\%$。
下面是一步胺交换反应。上一步得到的产物与 $\ce{NH3}$ 反应,结果相当于将其中一个 $\ce{NHAc}-$ 基团脱去保护:

随后是两步氧化反应,第一步是使用四氧化锇($\ce{OsO4}$)将双键双羟化,随后使用 $\ce{NaIO4}$ 氧化邻二醇,中间的碳碳键断裂:
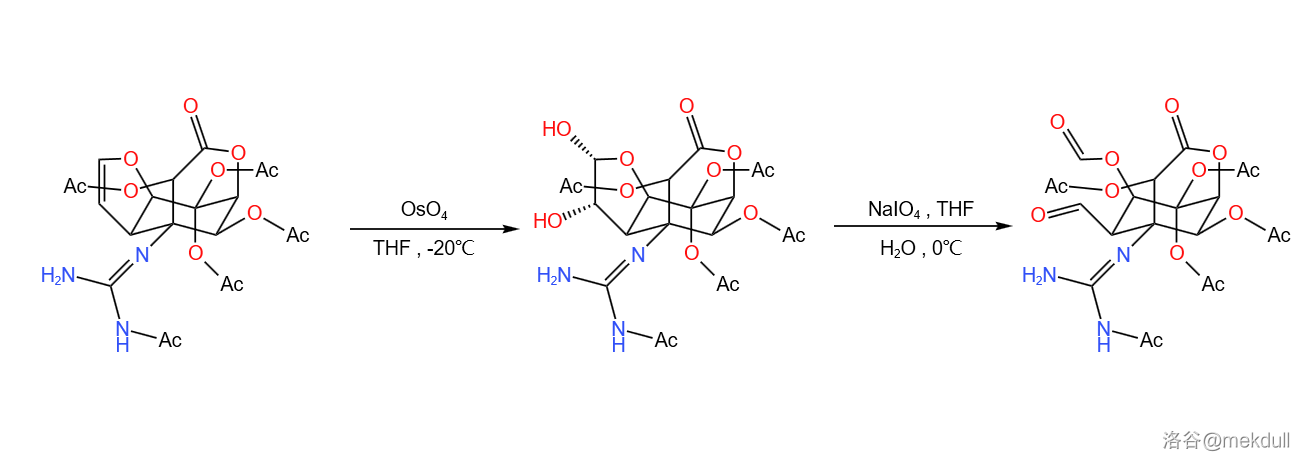
两步氧化反应的机理如下:

(上图:$\ce{OsO4}$ 氧化双键的机理)
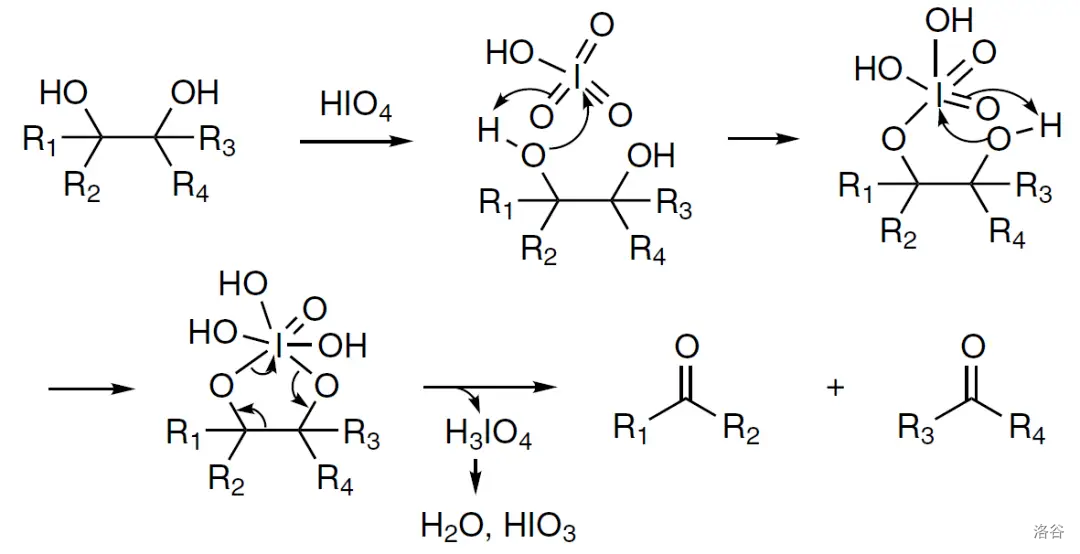
(上图:$\ce{NaIO4}$ 断裂邻二醇的机理)
最后一步,再弱碱性条件下,左侧的氨基($\ce{NH2}-$)对醛羰基亲核进攻,环化后脱去所有乙酰基保护,得到最终产物:
三步反应总产率 $25\%$。(可以看到,随着分子结构的愈发复杂,反应的产率也开始降低,这还算是比较正常的)。现在,我们完成了外消旋河豚毒素的全合成,总产率 $0.5\%$。
4.总结与后记
$1972$ 年,$Kishi$ 课题组对外消旋河豚毒素的合成是一项卓越的工作。它在有机合成化学历史上的意义远远超过了这种分子本身,后续的科学家在这项工作的基础上借鉴到了许多合成复杂笼状化合物的经验与方法。他们对立体化学、空间结构的控制令人叹为观止,整条合成路线简直就是一种令人赏心悦目的艺术。
但美中不足的是,$Kishi$ 课题组的路线并没有实现河豚毒素的不对称合成。一直要到 $2003$ 年,$Du~Bois$ 领导的课题组才完成了不对称合成。为此,他们专门发展了 $Rh-N$卡宾插入的 $C-H$ 键活化方法($Du’s~C-H$ 键活化偶联)。他们的逆合成设计如下:
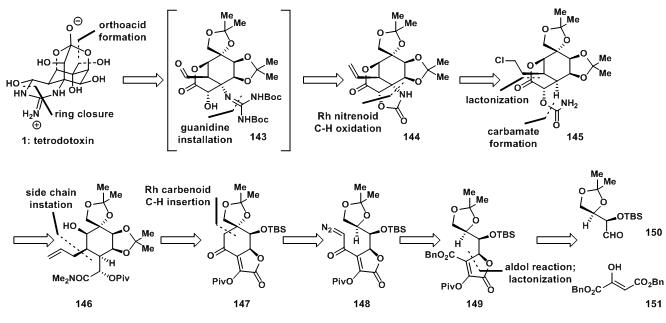
现在,这种合成方法已被推广,在其他物质的合成中也有了重要的应用。
但实际上,河豚毒素的全合成远不止这两例。几十年来,无数科学家前仆后继,他们的设计或许有好有坏,有些成功,也有些失败,但不管怎样,他们都是在这条路上的探索者。随着合成路线的不断优化,我相信,人们真正掌握并大规模合成这种物质的时代已在眼前。到时候,它将不再是令人生畏的毒药,而会以一种全新的面孔出现,造福了芸芸众生,也实现了它自己的涅槃。或许这,就是有机合成一直这么吸引我的原因吧。