谨以此文,纪念维生素B12被人类发现90周年。
注:如果本文中有任何错误或疏漏,请务必联系作者或在评论中写出。不胜感谢!
1.前情提要
维生素B12($\text{Vitamin~B12}$,又名钴胺素,缩写 $\text{VB12}$),这种物质(其实严格来说是一类物质)如今早已家喻户晓。作为唯一一种含有金属元素的维生素,其中心的 $Co$ 原子赋予了它鲜红的色彩,也让它有了一个别称:红色维生素。

巧合的是,鲜红的维生素B12最重要的作用之一,就是为我们的身体制造鲜红的血液。具体来说,维生素B12是一种辅酶,参与红细胞的制造与成熟,也参与某些氨基酸(如蛋氨酸)和胸腺嘧啶在体内的合成。总之,它在人体内发挥着非常重要的作用,是维持正常代谢和机能不可或缺的物质。
同时,维生素B12是也结构最复杂的维生素。它的化学式为 $\ce{C63H88N14O14PCo}$,分子量高达 $1355.37$,拥有一个咕啉($corrin$)大环(注意它与卟啉结构的区别)、一条结构类似于核苷酸的长链、$14$ 个具有手性的碳原子和一个在中心配位的钴原子,如下图:
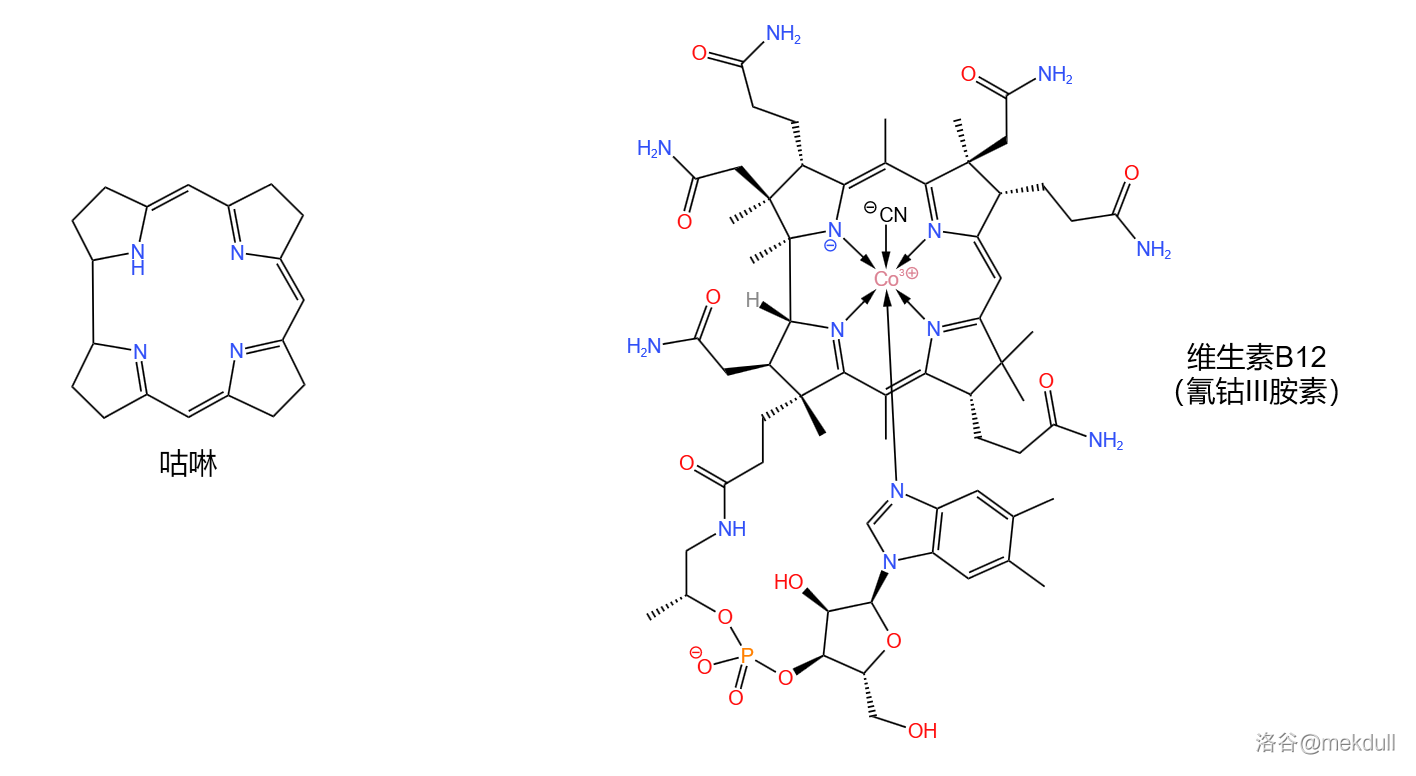
(注:严格来说,氰钴$\text{(III)}$胺素只能算是维生素B12的一种,但一般可以把它当做维生素B12的代表。下文中所有“维生素B12”字眼基本都指代这一物质。)
虽然维生素B12对正常生理活动非常重要,但是高等动植物却不能自行制造维生素B12。自然界中,这种物质主要依靠细菌等微生物供给,而我们则必须依靠摄食来补充。
人类对维生素B12的认识是漫长而曲折的。$19$ 世纪 $50$ 年代,英国医生 $Addison$ 首次描述了一种致死的恶性贫血,并发现这种疾病与患者的胃部受损有关。之后的几十年间,人们一直在试图搞清楚这种病的根源,但进展寥寥。直到 $1929$ 年,美国科学家 $Castle$ 提出了著名的双因子理论来解释发病机制:病人由于胃部受损,缺乏一种内因子($intrinsic~factor$),导致其无法从食物中吸收一种外因子($extrinsic~factor$),进而导致患病。
刚开始,许多科学家都对这套理论不以为然。直到 $1934$ 年,$G.~Whipple$、$G.~Minot$ 和 $W.~Murphy$ 三人真的发现且分离出了这种“外因子”,并将其取名为维生素B12,对应的争论才偃旗息鼓。三人也因为这项重大成果获得了诺贝尔生理学或医学奖:

(上图:三位发现维生素B12的科学家)
$1948$ 年,$Rickers~E.~L.$ 等人得到了纯的维生素B12。随后,对其复杂结构的破译轰轰烈烈地展开。$8$ 年之后,$1956$ 年,$D.~Hodgkin$ 等人用X射线衍射法彻底破解了它的结构,并因此获得了 $1964$ 年的诺贝尔化学奖。
好,既然结构已经确认,那么下一步当然就是合成这种物质。可是,它的结构是如此巨大而复杂,简直令人望而生畏。当时的化学家们都将其称为有机合成界的珠穆朗玛峰。(虽然后面又出现了更加复杂的物质(Link:岩沙海葵毒素的全合成)拿走了这一称谓)
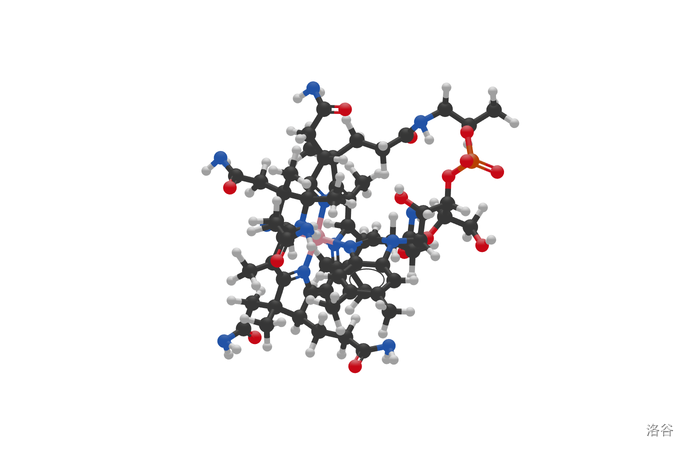
(上图:维生素B12的球棍模型,其中黑色:$C$,白色:$H$,红色:$O$,橙色:$P$,蓝色:$N$,粉色:$Co$)
但是,人类并不会因此放弃。
从 $1959$ 年开始,瑞典化学家 $A.~Eschenmoser$ 等人就开始致力于攀登这座高峰,成为了这项工作的先行者。$1960$ 年,德国化学家 $Bernhauer~K.$ 等人加入了攀登者的行列,并成功完成了从钴啉胺酸($cobyric~acid$)到维生素B12的半合成。$1961$ 年,刚刚完成叶绿素A全合成(Link:叶绿素A的全合成)的“有机合成艺术家”$R.~Woodward$ 带领着他的课题组加入了进来,并很快与其他研究者取得合作。最终,$1972$ 年,在整整 $13$ 年的光阴之后,这项超过 $100$ 位研究者参与的、跨越半个地球的“有机合成化学史上最疯狂的工作”终于宣告成功。

(上图:《维生素B12的全合成》论文影,$1973$,伍德沃德。参与工作的其他课题组也发表了各自的论文。)
在下文中,我们将会详细讨论这条凝聚了上百人心血的维生素B12全合成路线。不要因为它的复杂而感到畏惧,请相信我,当你逐渐理解它的时候,你会发现它的精巧和美丽。它是一件巧夺天工的艺术品。
在上帝创造的自然界的旁边,化学家们又创造了一个新的世界。——伍德沃德,$1956$
2.概述
与叶绿素A一样,维生素B12也是一个相当脆弱的分子。它只有在 $\text{PH}=4.5-5.0$ 的狭小区间内才能长时间稳定,而一旦 $\text{PH}>7$ 或是 $\text{PH}<2$ 就会迅速分解。它的热稳定性也不好,在 $100^{\circ}C$ 以上也会彻底分解。这无疑是给全合成工作带来了巨大的挑战。
但是,这还并不是合成过程中最大的难点。前面提到,维生素B12拥有一个咕啉大环(下图中紫色部分)——这是整个分子的核心结构。它由 $4$ 个氢化的吡咯环拼接而成,具有显著的不对称性。由于无法形成 $\pi$ 电子的环流,咕啉环没有芳香性,性质十分脆弱,且周边分布着众多的手性碳原子。当时的手性合成技术还远没有现在成熟,因此在维生素B12之前,人们还从来没有成功合成过咕啉衍生物。
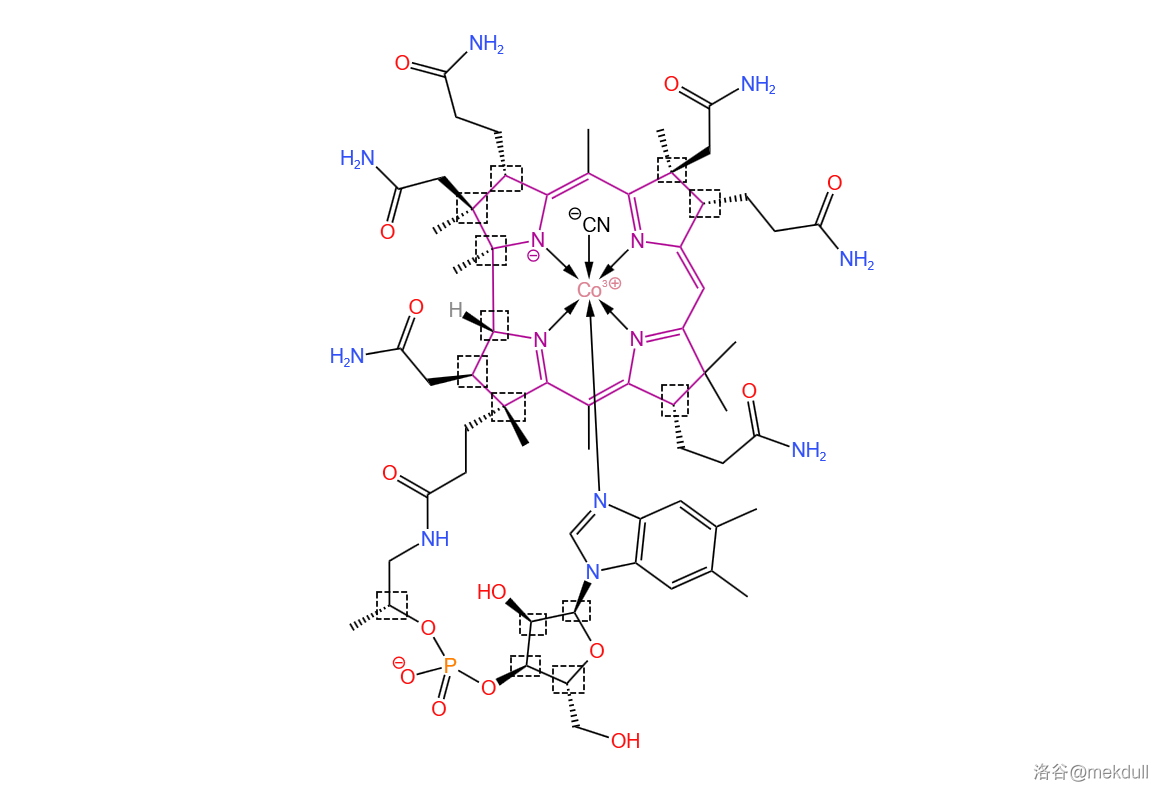
(上图中:紫色标出了咕啉结构;虚线框框出了所有手性碳原子)
$1960$ 年,分子下方的那条类似于核苷酸的长链被 $Bernhauer~K.$ 等人成功合成——这也算是全合成中的第一个阶段性成果。于是,后续合成工作的目标变成了钴啉胺酸(如下图),而咕啉结构的构建也随即成为了核心问题:

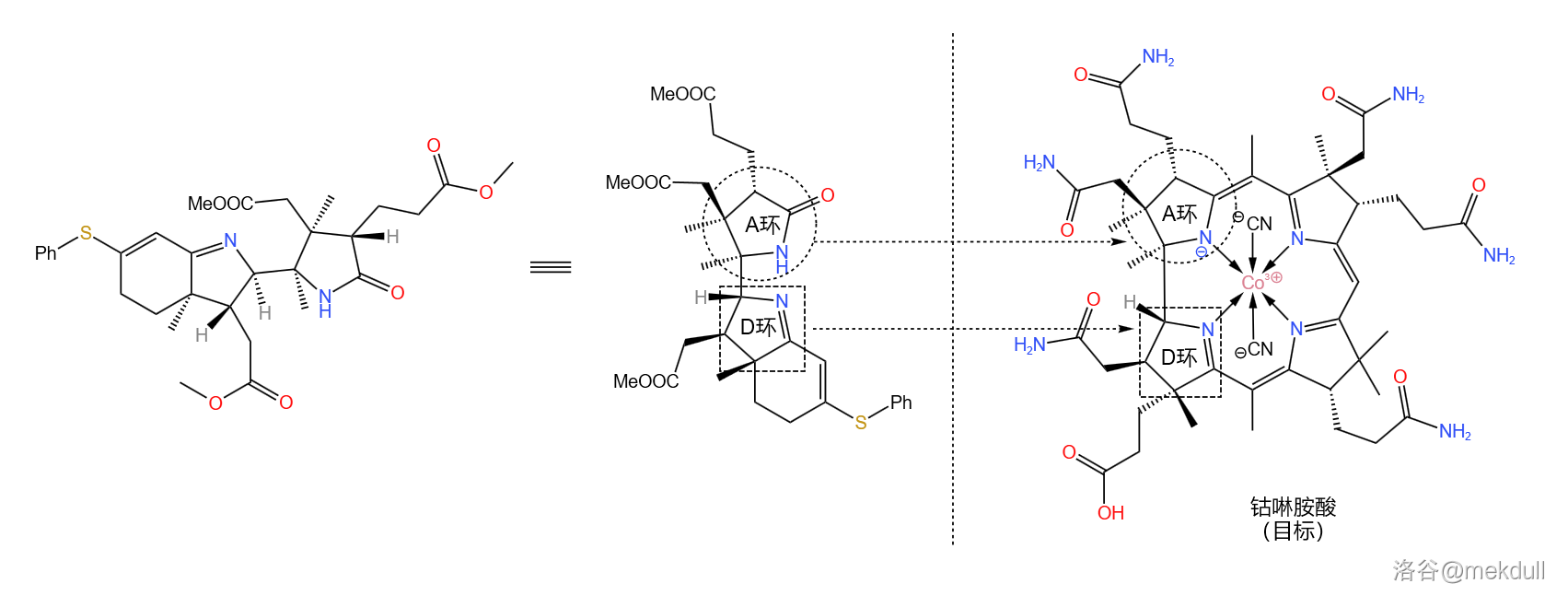
解决这一问题的思路是将整个分子劈成两半(如下图),然后再将它们拼接到一起。其中,左半部分被命名为 $cyanobromide$,由 $R.~Woodward$ 的团队负责合成;右半部分被命名为 $thiodextrolin$,由 $A.~Eschenmoser$ 的团队负责合成:
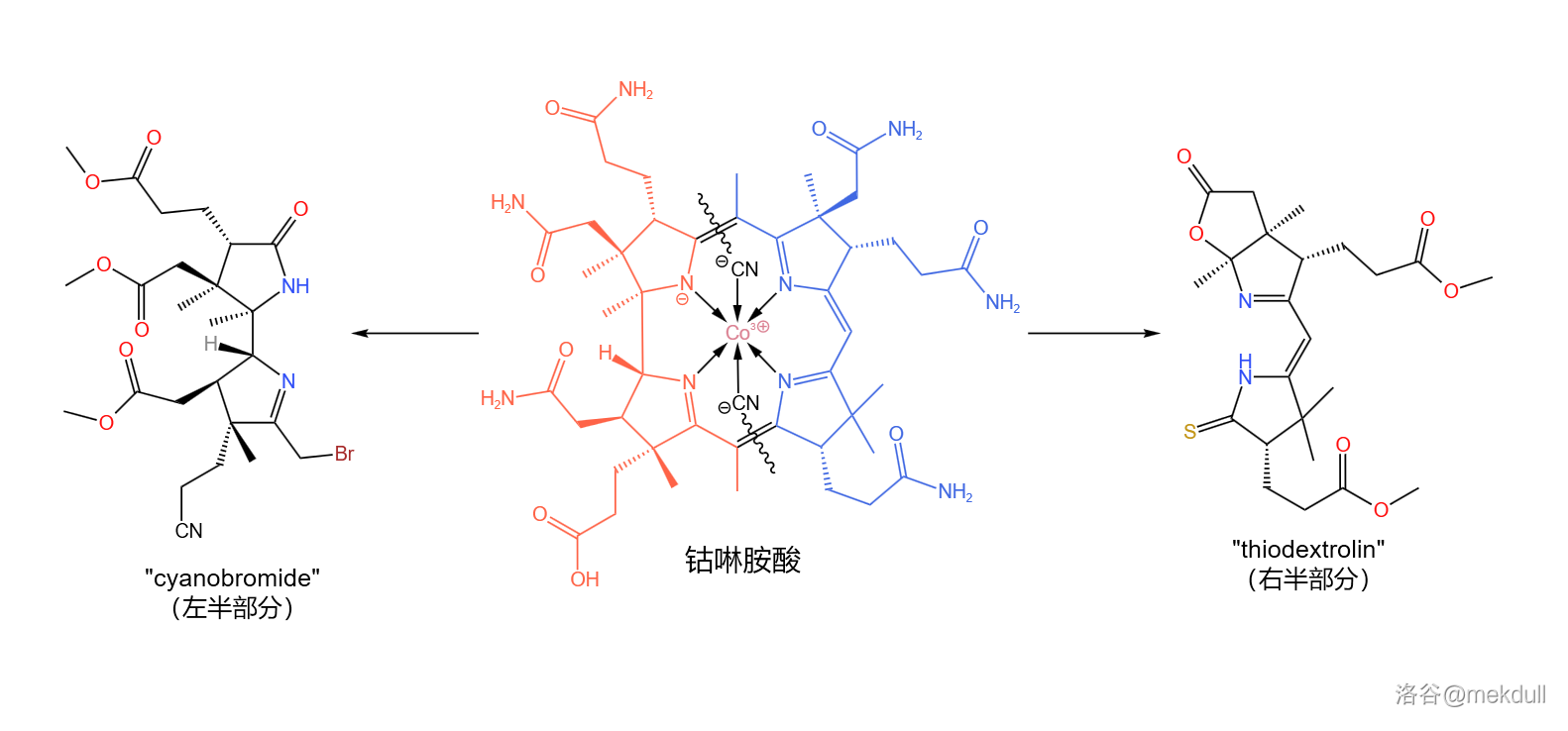
现在,分工已经确定,全合成的介绍也将正式开始。
本文中涉及的主要有机反应清单:
- $Kucherov$ 加成;
- $Wittig$ 反应;
- $Birch$ 还原;
- $Criegee$ 臭氧化反应;
- 羟醛缩合反应;
- $Beckmann$ 重排;
- $Diels-Alder$ 反应;
- $Arndt-Eistert$ 同系增碳反应;
- $Eschenmoser$ 偶联反应;
- $Eschenmoser$ 酰胺水解反应;
- $Meerwin$ 盐的烷基化反应。
3.逐步合成分析
3.1 左半部分的合成
左半部分的合成起始于一种普通的吲哚衍生物——$6-$甲氧基$-2,3-$甲基吲哚——虽然现在它看上去似乎与目标完全不沾边。首先,用格式试剂(这里使用的是甲基碘化镁,$\ce{MeMgI}$)与溴丙炔与它反应,在 $3$ 号位上接上一个炔丙基:
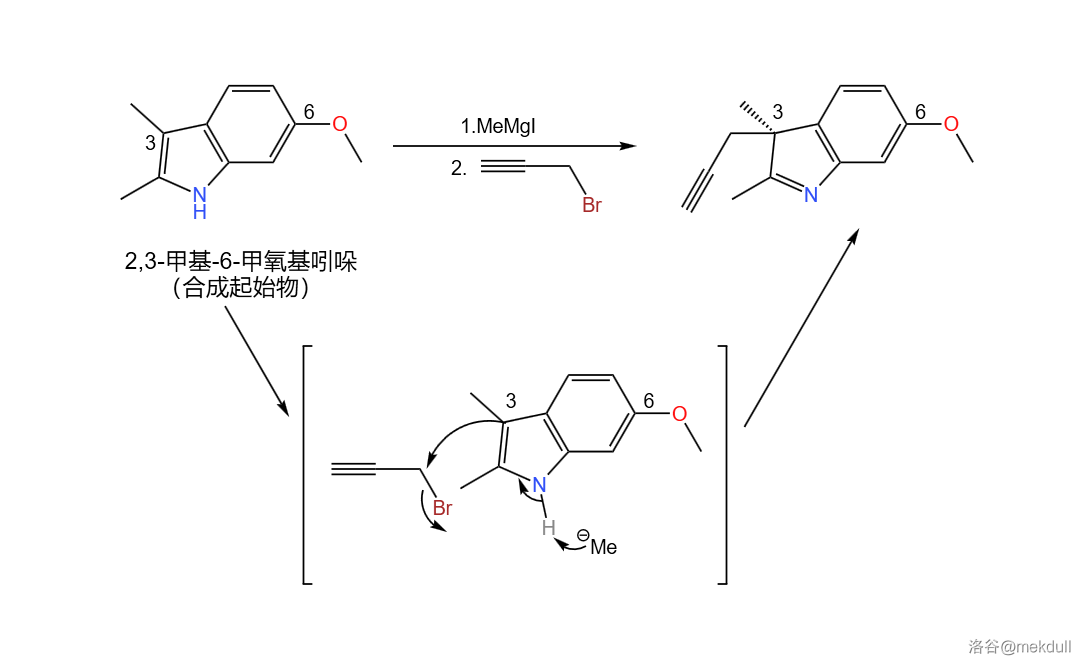
实际上,这个反应的立体选择性并不好,得到的产物是外消旋体,在后续的步骤中会将其分离。
下一步,在路易斯酸(此处使用 $\ce{BF3}$)与 $\ce{Hg(II)}$ 物种的催化下,碳碳叁键与甲醇发生著名的 $Kucherov$ 加成反应,得到烯基醚;随后,分子内自发环化,最终得到一个三环结构的酮:
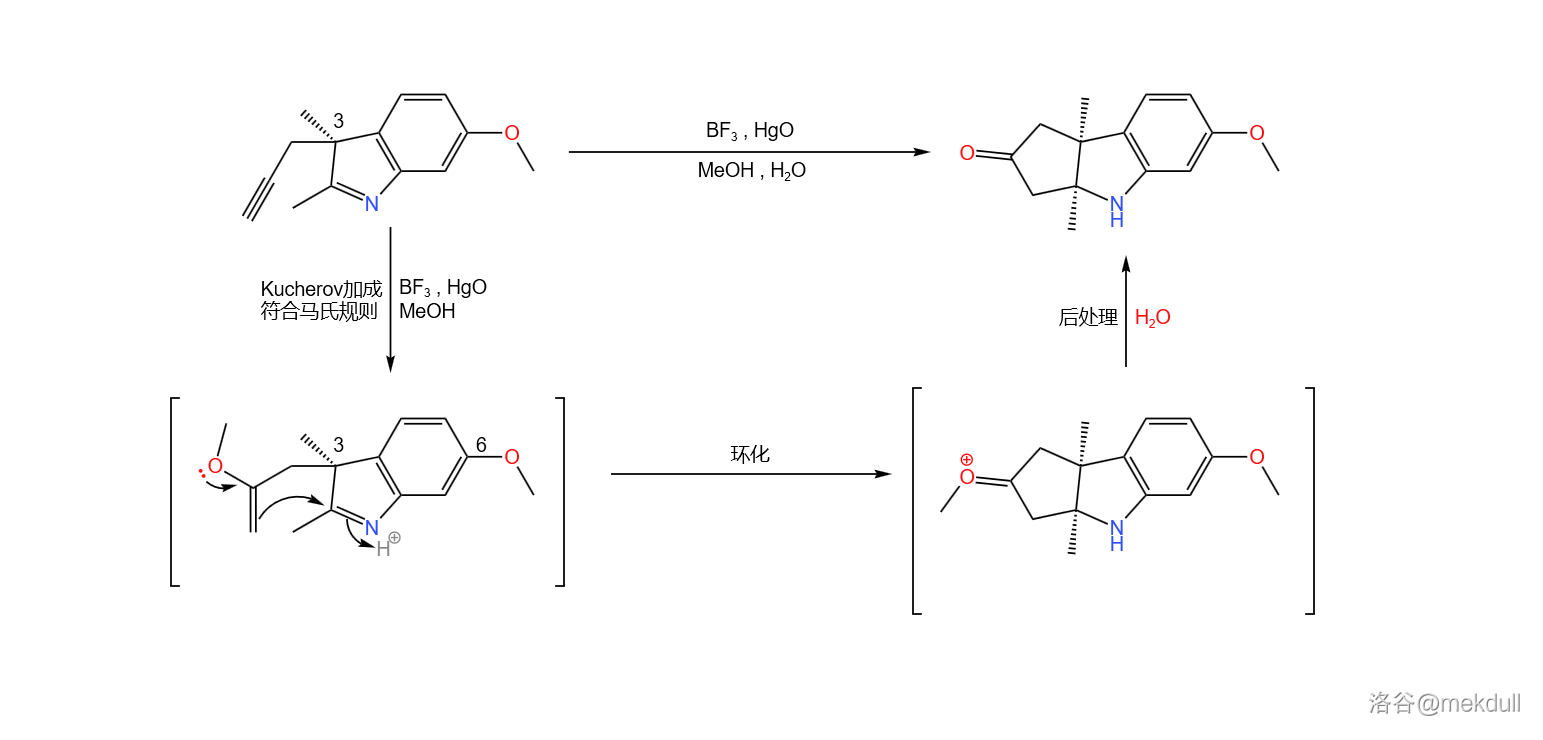
(注:由于 $3$ 号位有甲基的存在,导致有一侧的进攻受阻,因此产物的立体构型是单一的。)
现在,我们先把产物放在一边备用,来处理第二个合成起始物。它被称作樟烯酸甲酯($methyl~laurolenate$,见下图),是樟脑经过一系列反应后得到的产物,很早之前就被人们合成。伍德沃德之所以选择它,主要是因为它拥有一个继承自樟脑的手性碳原子。
首先,用氢化铝锂($\ce{LiAlH4}$)处理 $\text{(S)}-$樟烯酸甲酯,将其还原为醇;随后再用铬酸($\ce{H2CrO4}$)氧化,得到一种醛;最后,使用著名的 $Wittig$ 反应得到一个碳碳双键:
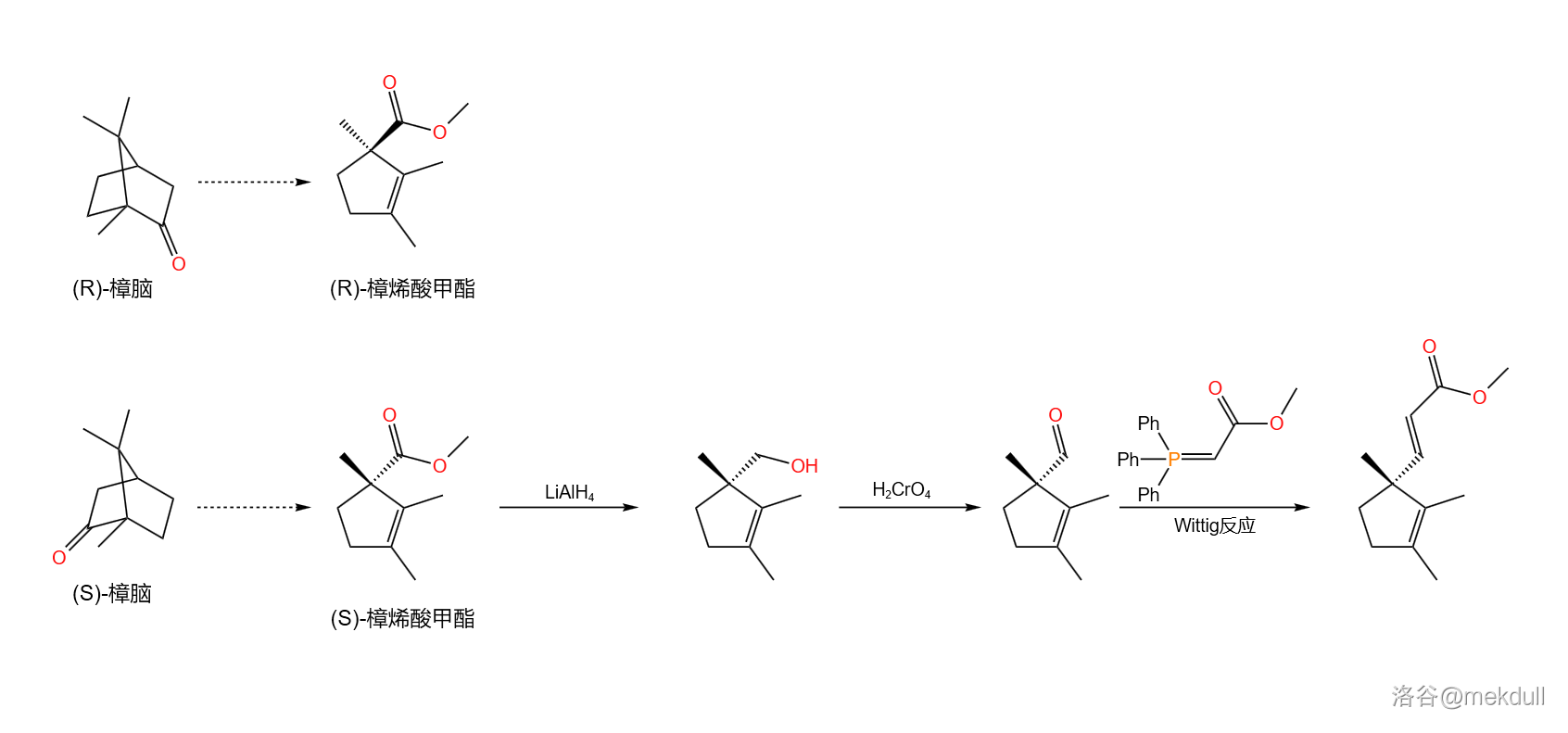
$Wittig$ 反应是一个重要的烯烃合成反应,其机理在之前的几篇文章中都有提及,在此便不再赘述。
接着,我们用酸将得到的酯水解,再用氯化亚砜($\ce{SOCl2}$)将其变为酰氯。酰氯随即与刚才合成的那个三环结构的酮反应,两者结合在一起形成酰胺结构:
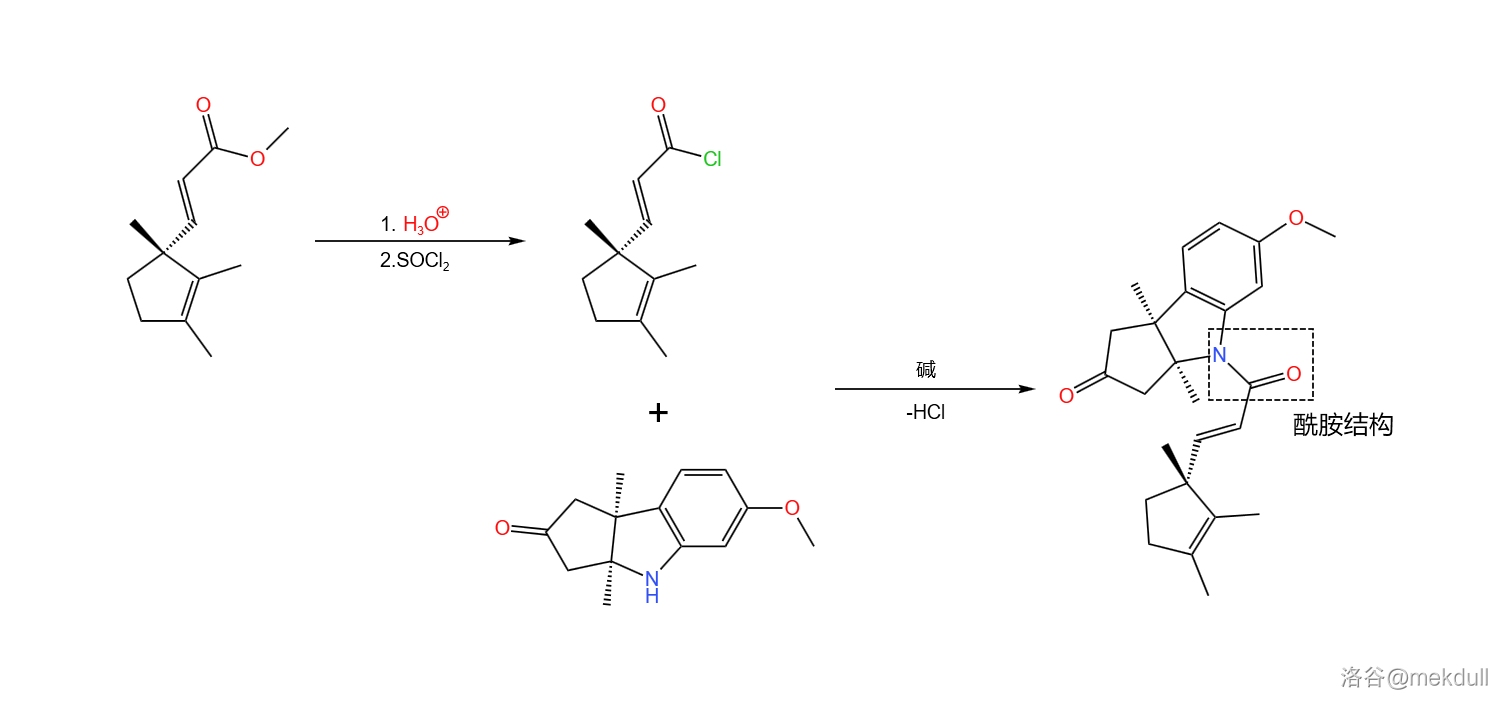
随后是一个精彩的环化反应:在丁醇钾($\ce{t-BuOK}$)的作用下,$19$ 号碳原子上的氢原子被夺去,形成烯醇负离子;烯醇负离子进攻附近 $\alpha,\beta-$不饱和的羰基环化。由于分子的构型已经固定,进攻只有一个方向,因此得到了单一立体构型的产物:
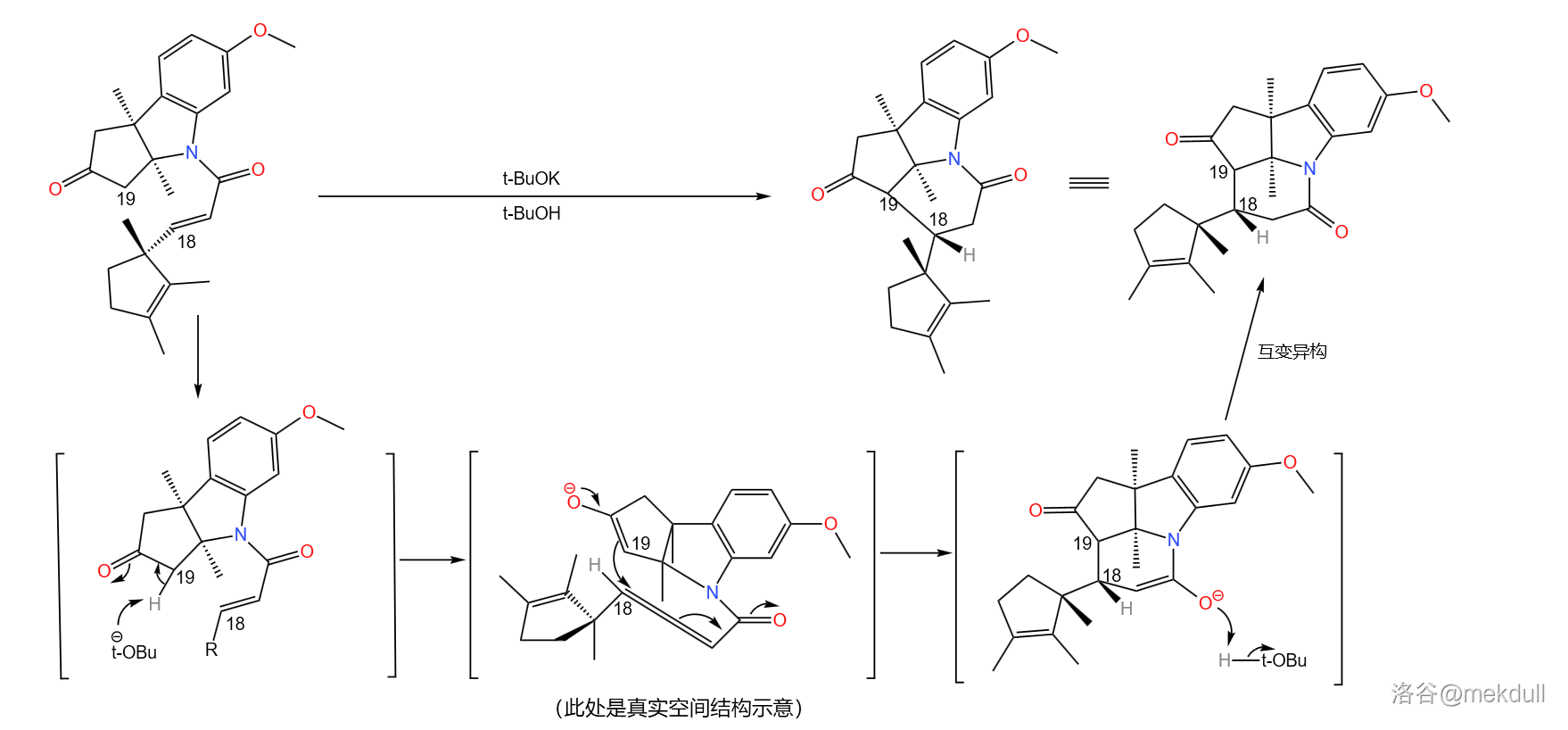
(注:这里的编号表示了该碳原子最终在咕啉环中的位置)
接下来,用乙二醇($\ce{HO(CH2)2OH}$)保护 $2b$ 号碳原子上的酮羰基。由于分子中还有一个酰胺结构(位于 $18b$ 号碳原子),所以我们还得用 $Meerwin$ 盐再次进行保护(注:酰胺中的羰基无法被乙二醇保护):
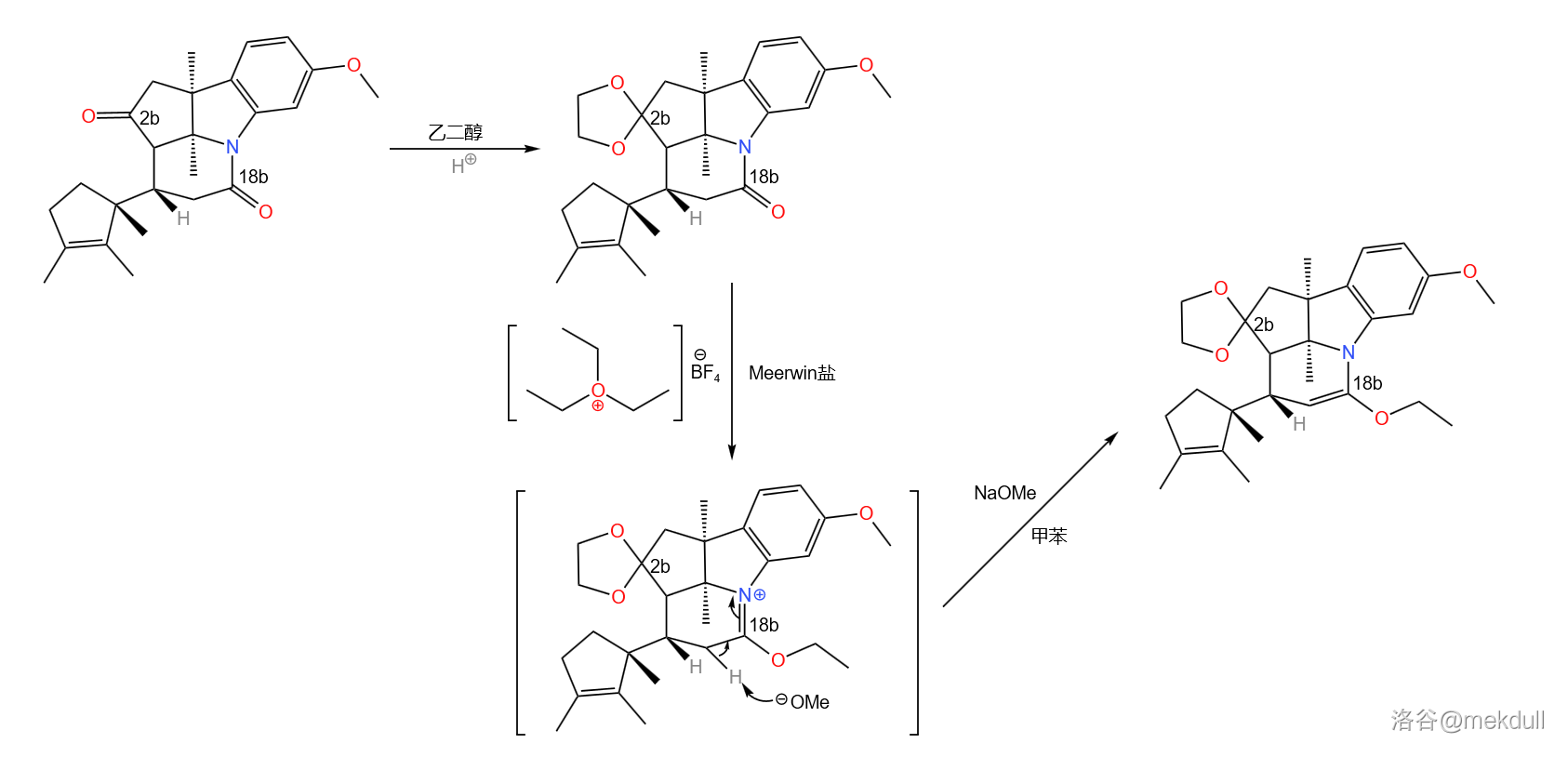
之所以要这样做,是因为下一步要用到一个比较强烈的反应,即著名的 $Birch$ 还原反应。在液氨中,用过量的碱金属(这里使用 $Li$)处理这一物质,右上方的苯环被还原为环己二烯结构;随后用酸处理之,经过双键移位后得到 $\alpha,\beta-$不饱和酮;同时,上一步的保护也被脱去:
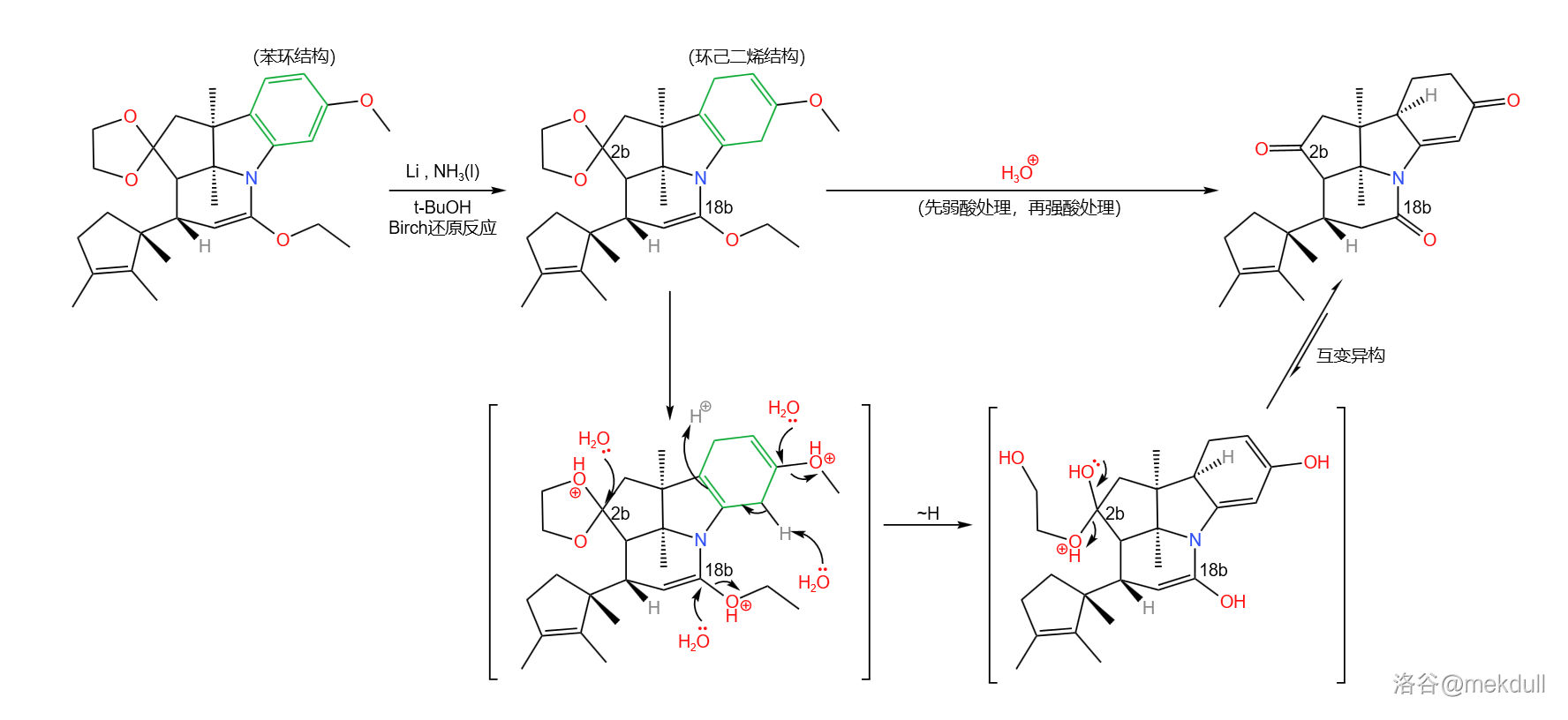
$Birch$ 还原是非常经典的自由基还原反应,在有机化学史上具有重要的地位。反应机理如下图所示:
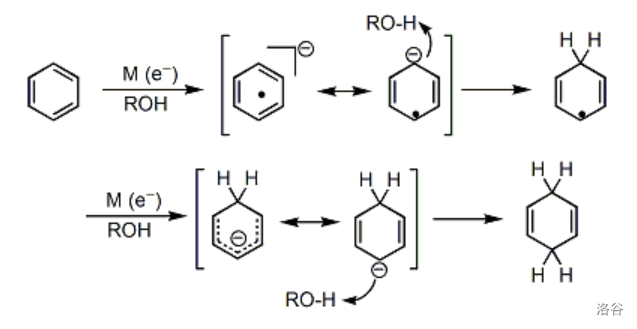
显然,对于带有取代基的苯环来说,最终生成的烯烃的位置选择性,是由中间体的稳定性决定的。一般来说,连接着吸电子基团($\text{EWG}$)的碳原子最终不会带有双键,而连接着给电子基团($\text{EDG}$)的碳原子最终会带有双键。例如:
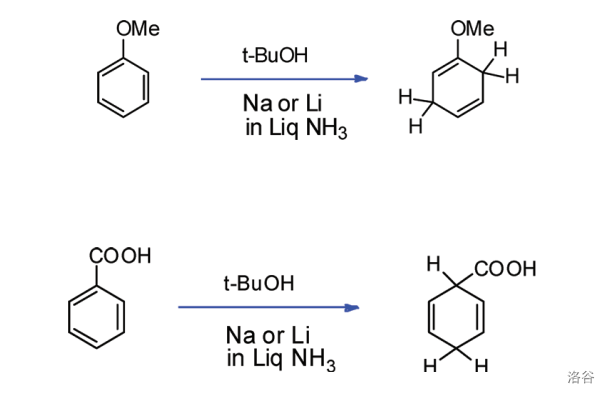
(注:想要更深入了解 $Birch$ 还原的位置选择性的读者请见参考文献 $[11]$)
如果具体到我们这个反应,你会发现:苯环的 $a,b,d$ 三个位置都带有给电子基团,所以还原产物是单一的。没错,这也是伍德沃德刻意的设计:
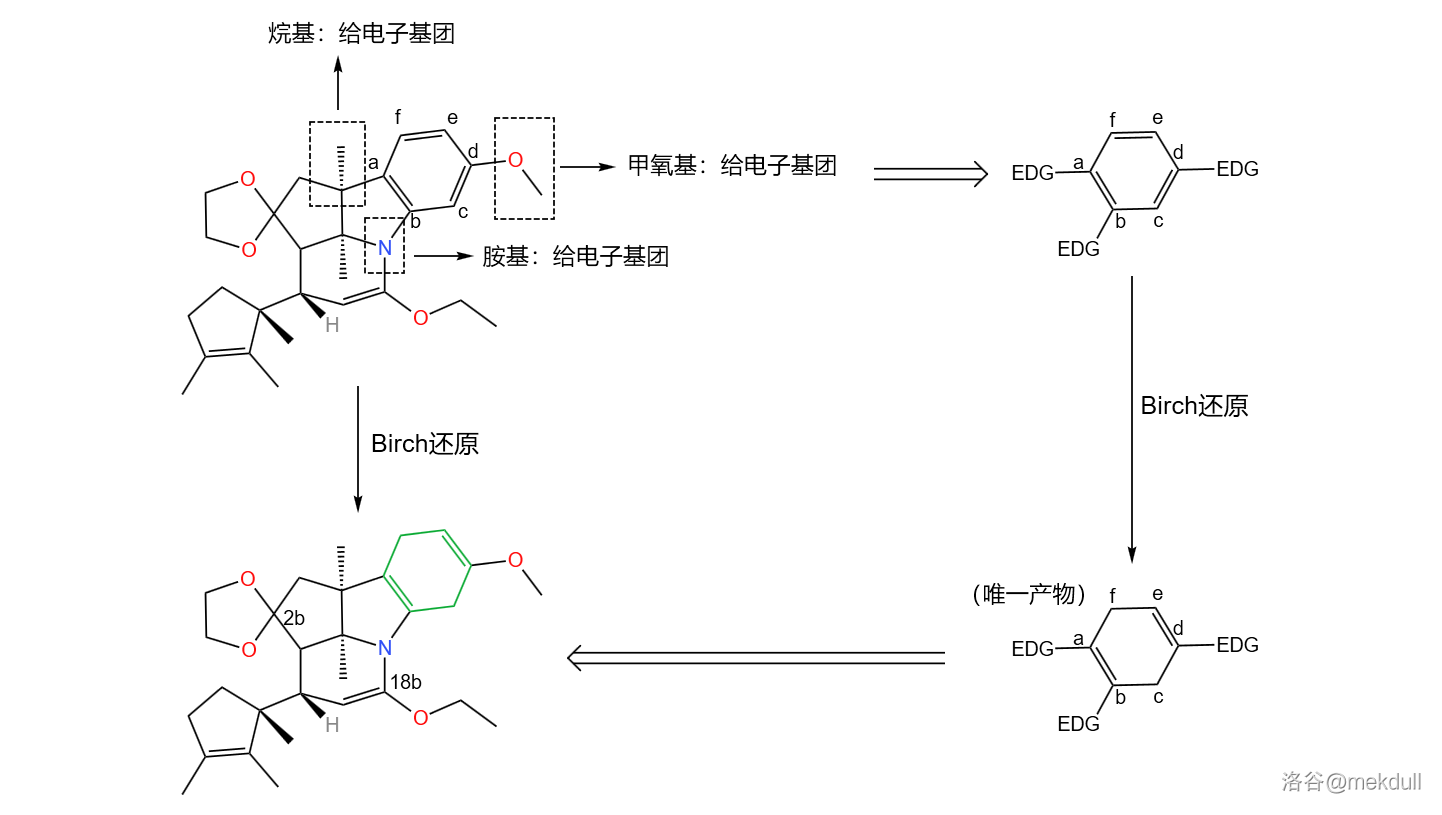
应该说,课题组的设计总体来说是成功的——两步反应的总产率达到了惊人的 $90\%$。但它们很快发现了一个问题:产物中 $3$ 号碳原子的构型与预料中的不符。
理论上,从化学动力学角度,由于附近甲基的空间位阻,$\ce{H}$ 原子应该结合在甲基的异侧;但从化学热力学角度,$\ce{H}$ 原子与甲基在同侧时能量更低,也就更加稳定。但实际是,我们得到的几乎全是热力学控制的产物——而那并不是我们想要的。课题组尝试改变反应条件,但都无济于事:
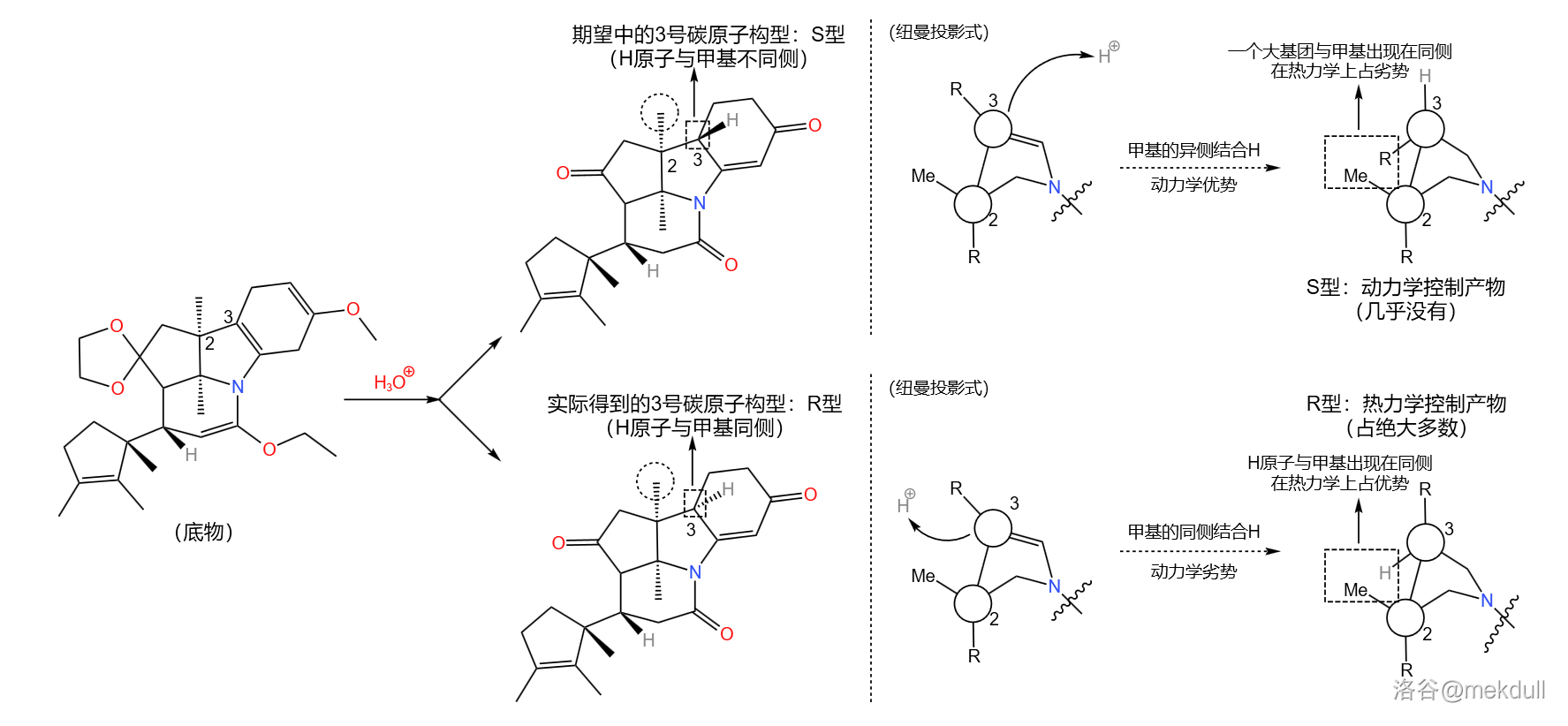
在关键时刻,伍德沃德表现出了超乎常人的冷静。经过分析,它认为:$3$ 号碳原子是特殊的,在特定条件(比如酸性或碱性条件)下,它可以通过互变异构的方式转换自己的构型(注:一般来说,构型固定之后不会轻易翻转,因为构型翻转需要断开化学键)。由于物质总是自发地趋向于稳定,所以并不是我们制不出 $S$ 型,而是 $S$ 型产物会迅速变成在热力学上更稳定的 $R$ 型:

既然如此,我们是不是也可以利用这一特性,把构型翻转回来?只需要通过某种手段,让 $S$ 型比 $R$ 型在热力学上更稳定就行了。于是,他在论文中写道:
我们相信,如果他能在我们不需要的时候大量生成,那么至少在原理上,对它进行必要的翻转不是一件困难的事情。——《在天然产物全合成中的最新进展》,伍德沃德,$1968$
他决定,暂时先不管这个构型问题,继续原有的计划。
下一步,用羟胺($\ce{NH2OH}$)处理产物,把分子中的两个羰基都变成肟。事实上,只有 $2b$ 号碳原子上的肟是我们需要的,但是对于羟胺来说,两个羰基的活性差不多(即使其中一个羰基是 $\alpha,\beta-$不饱和的),因此我们很难在这一步区分二者,只好先把它们全部变成肟:
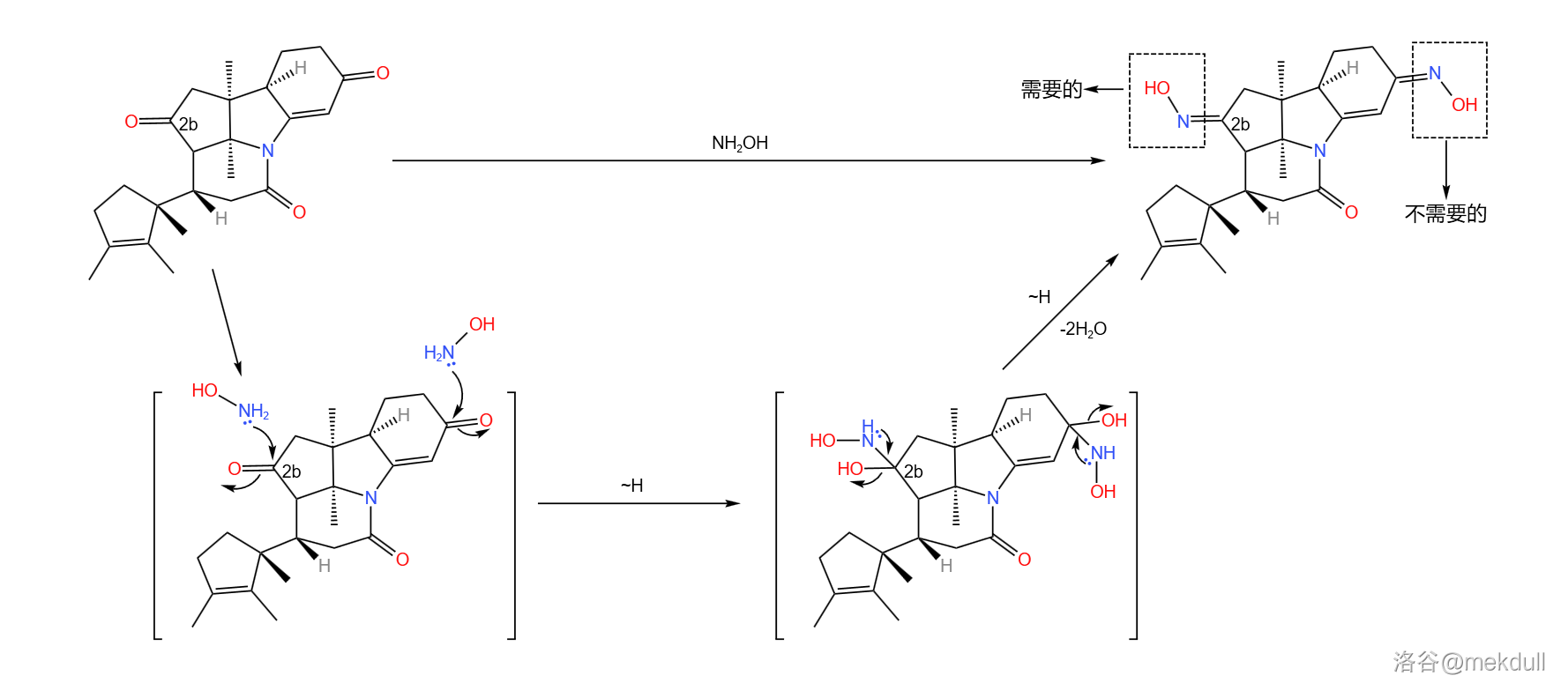
当然,我们还要把那个不需要的去掉。在非常温和的条件下,用一定量的亚硝酸($\ce{HNO2}$)与乙酸处理,可以把不需要的那个 $\alpha,\beta-$不饱和的肟基除掉,而 $2b$ 号碳原子上的肟基不受影响:
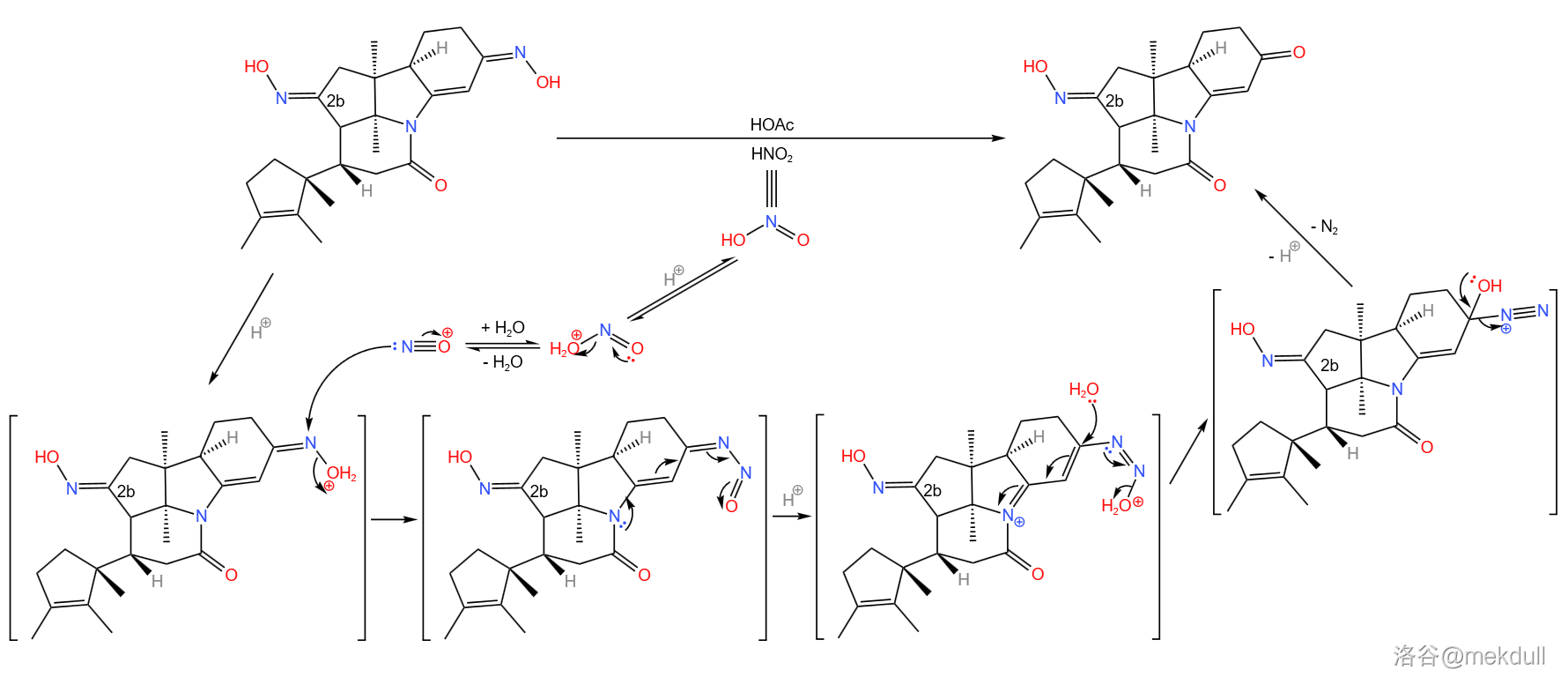
事实上,$2b$ 号碳原子上的肟基并不会马上用到,但后续的反应会产生越来越多的羰基,如果把这一步放到后面,那反应将更加难以控制。读者可以先猜一猜这个肟基后续会发挥什么作用。
回归正题,接下来登场的是著名的 $Criegee$ 臭氧化反应。在低温下用臭氧处理该物质,分子内的两个碳碳双键被打开,形成臭氧化物中间体;随后,加入高碘酸($\ce{H5IO6}$)氧化;最后,加入重氮甲烷($\ce{CH2N2}$)进行酯化:
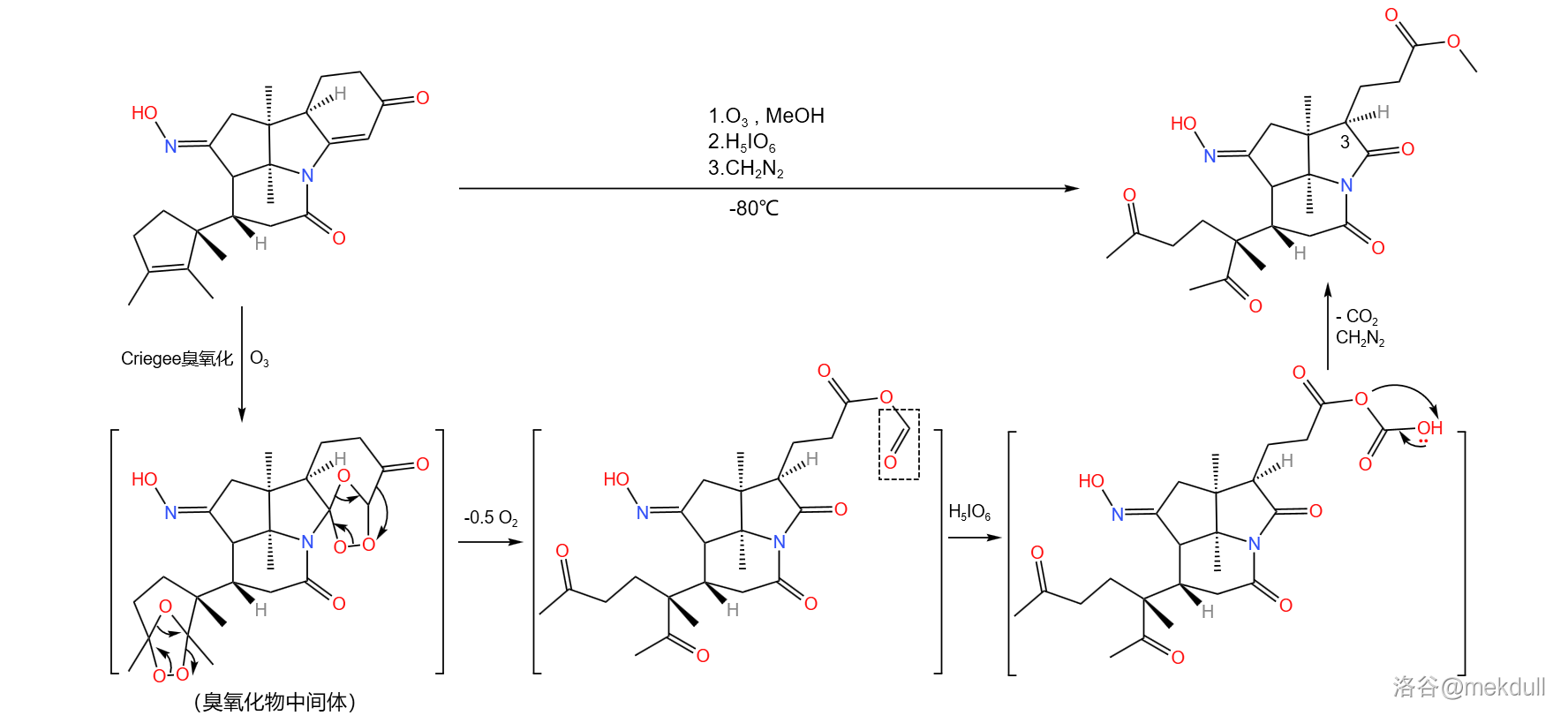
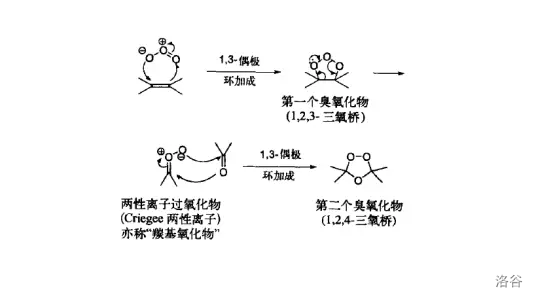
生成臭氧化物中间体的机理如下图所示。由于这一中间体很不稳定,所以 $Criegee$ 臭氧化一般需要在低温下进行。
接下来,在四氢吡咯的催化下,分子左下角发生经典的羟醛缩合反应,得到 $\alpha,\beta-$不饱和酮的结构;随后,用甲磺酰氯($\ce{MsCl}$)处理,将 $2b$ 号位上的肟基保护起来;最后,再来一遍 $Criegee$ 臭氧化,双键断裂,得到 $17c$ 号位的酯结构:
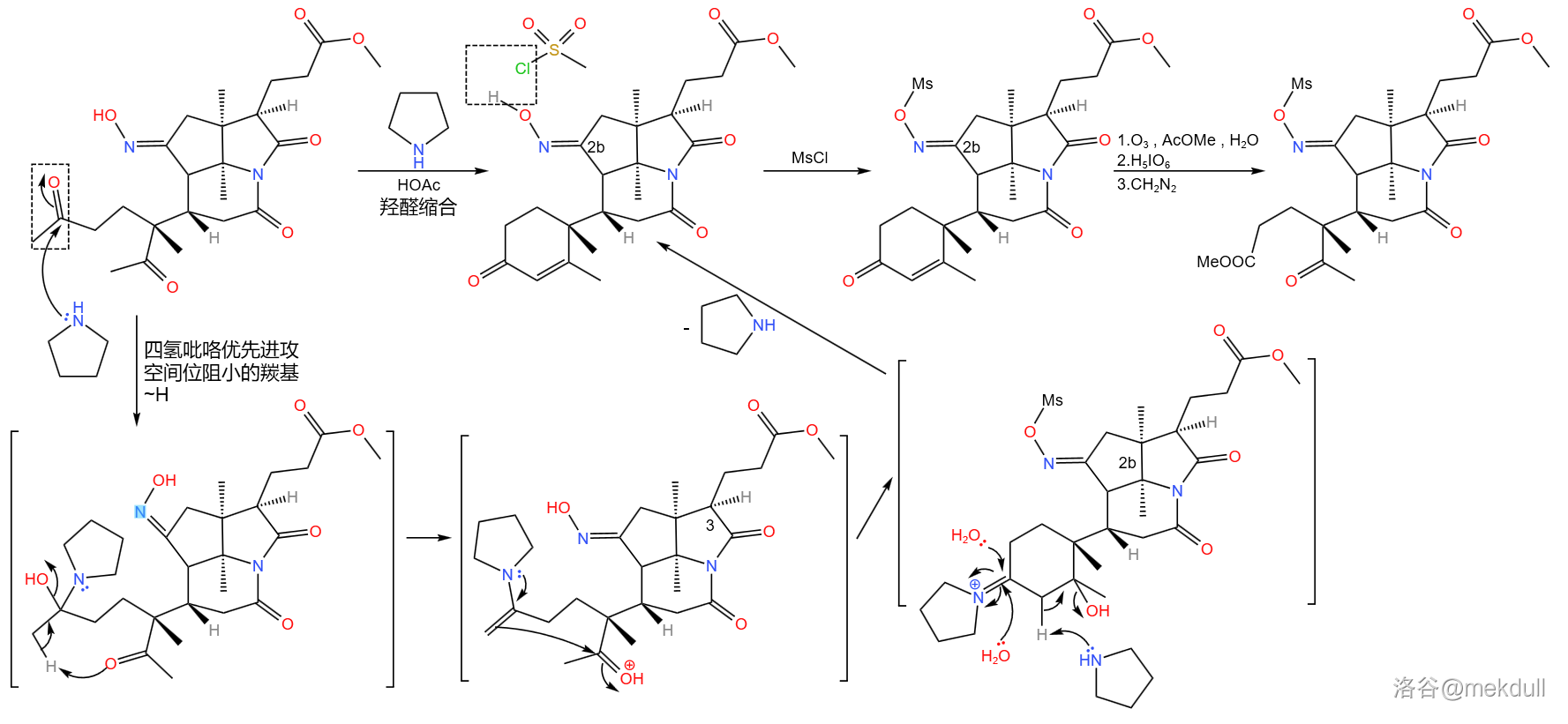
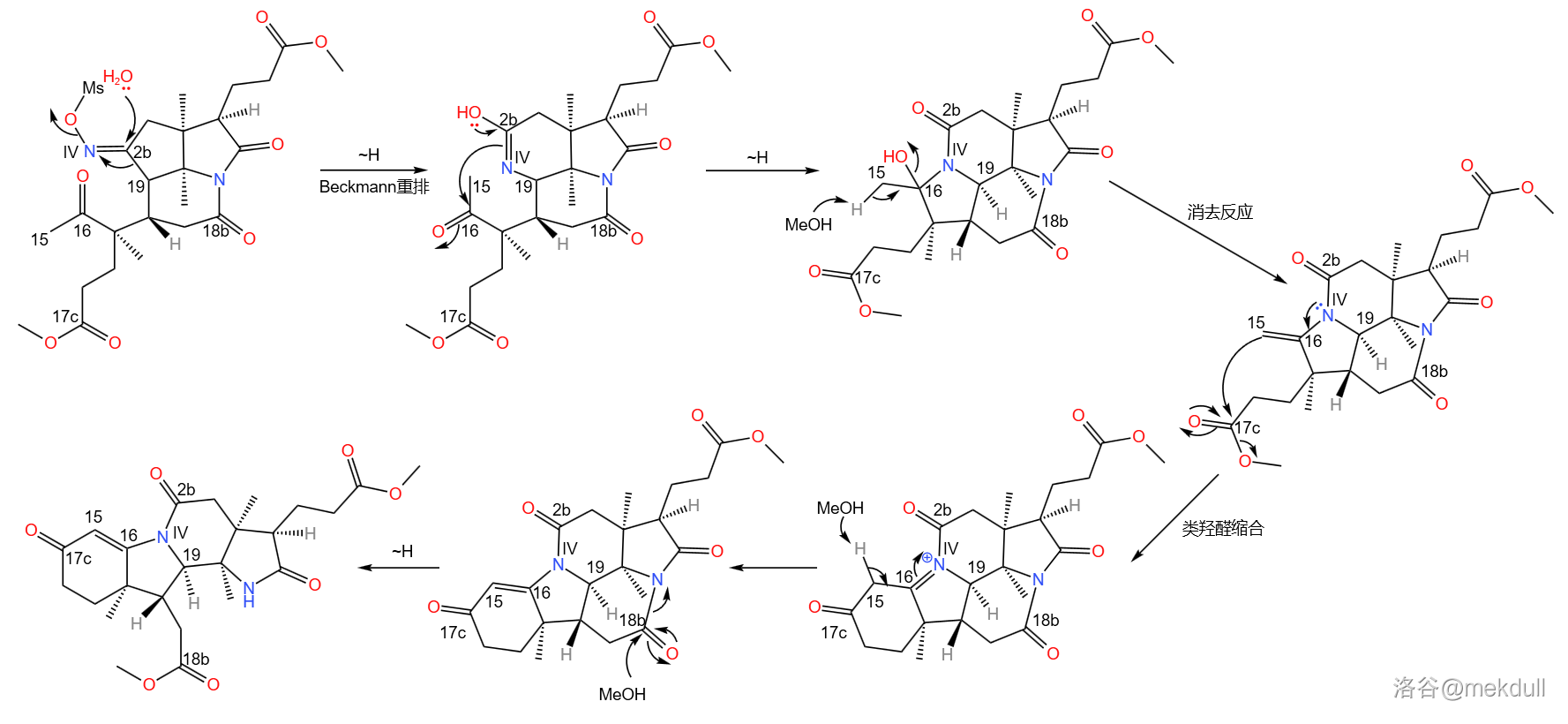
之所以如此大费周章,是因为伍德沃德准备要下一局大棋。现在,$2b$ 号位上的肟基终于真正要出场了——它将引导著名的 $Beckmann$ 重排反应。在 $170^{\circ}C$ 下,用聚苯乙烯磺酸($\ce{(C8H8O3S)_n}$,缩写 $\ce{PSS}$)和甲醇处理之,首先发生碳骨架重排,$19$ 号碳原子与 $2b$ 号碳原子分离;随后,$\ce{IV}$ 号氮原子亲核进攻附近 $16$ 号碳原子的羰基,形成一个五元环;接着,中间体消去 $1$ 分子水,形成 $15,16$ 号碳原子上的双键,从而形成烯胺结构;最后,烯胺亲核进攻附近 $17c$ 号碳原子上的酯基,在类似于羟醛缩合的反应后形成一个六元环状的 $\alpha,\beta-$不饱和酮结构。与此同时,在 $18b$ 号位上,原来的内酰胺环被打开,从而完成整个反应历程:
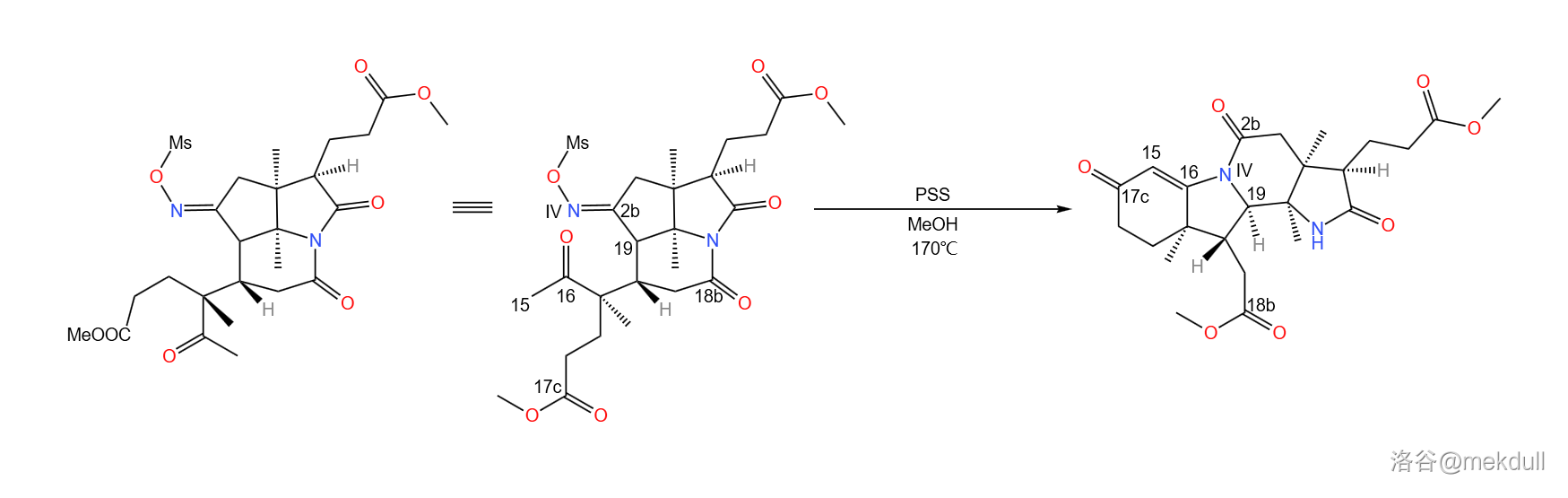
或许你已经在感叹这个反应的巧妙,但相信我,精彩并未结束。我们得到的产物被命名为半咕降甾酮($corrnorsterone$,关于这个名字有兴趣的可以看下面的“说文解字”)。还记得 $3$ 号位那个错误的构型吗?课题组发现,在一系列反应后,居然有一小部分产物的构型又翻了回来。也就是说,我们实际得到的是两种对映异构体的混合物。为了便于区分,课题组在它们的名字前加上了 $\alpha-$ 和 $\beta-$ 的前缀,如下图:
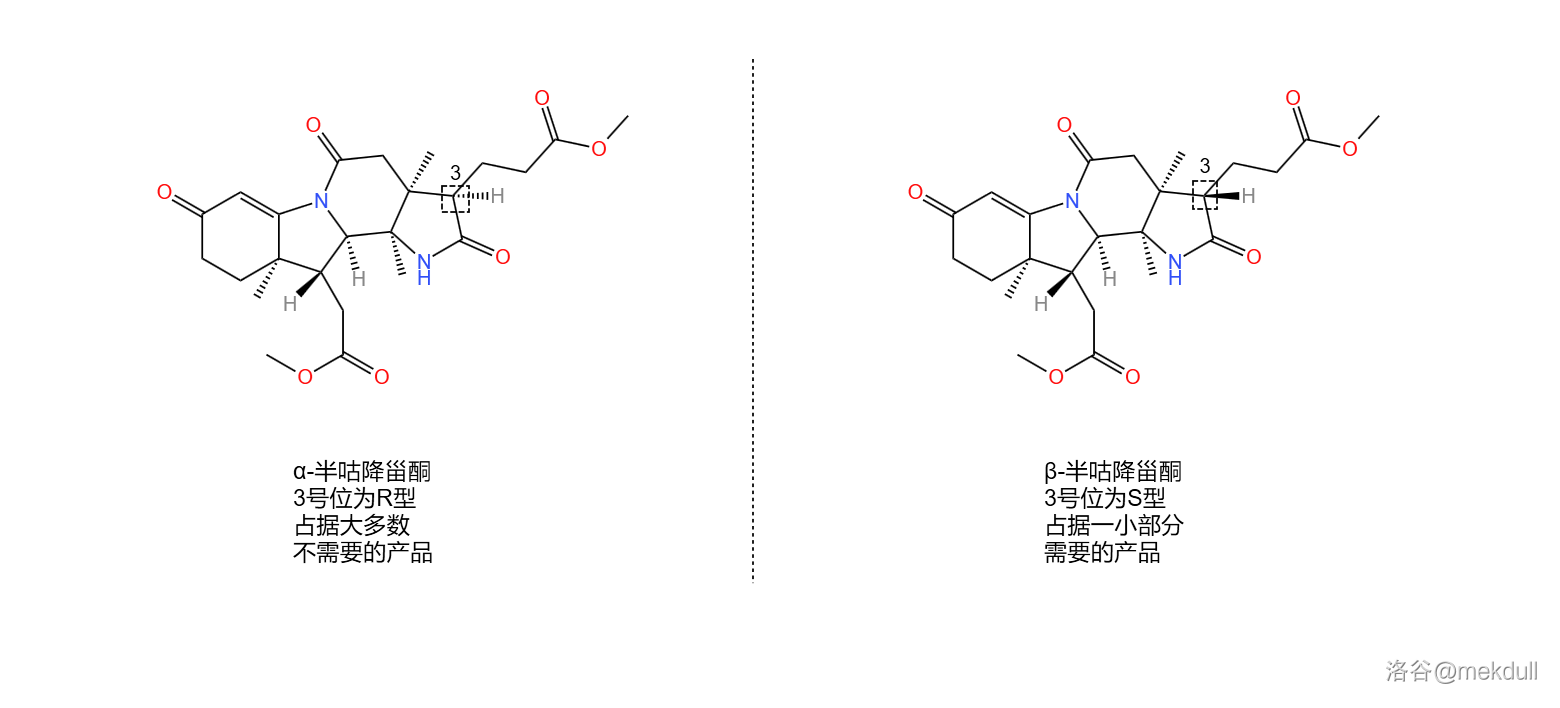
说文解字:$\text{corr-norster-one}$(半咕降甾酮)
“$corr$”是单词 $corrin$(咕啉)的一半,表示这种物质最终会变成咕啉环的左半部分;
“$norster$”源自单词 $norsteroid$(降甾醇,也泛指去甲甾族化合物),表示如果把 $N$ 原子换成 $C$ 原子的话,那么这个物质就具有降甾醇的碳骨架;
“$one$”是酮类化合物的后缀,源自单词 $ketone$。
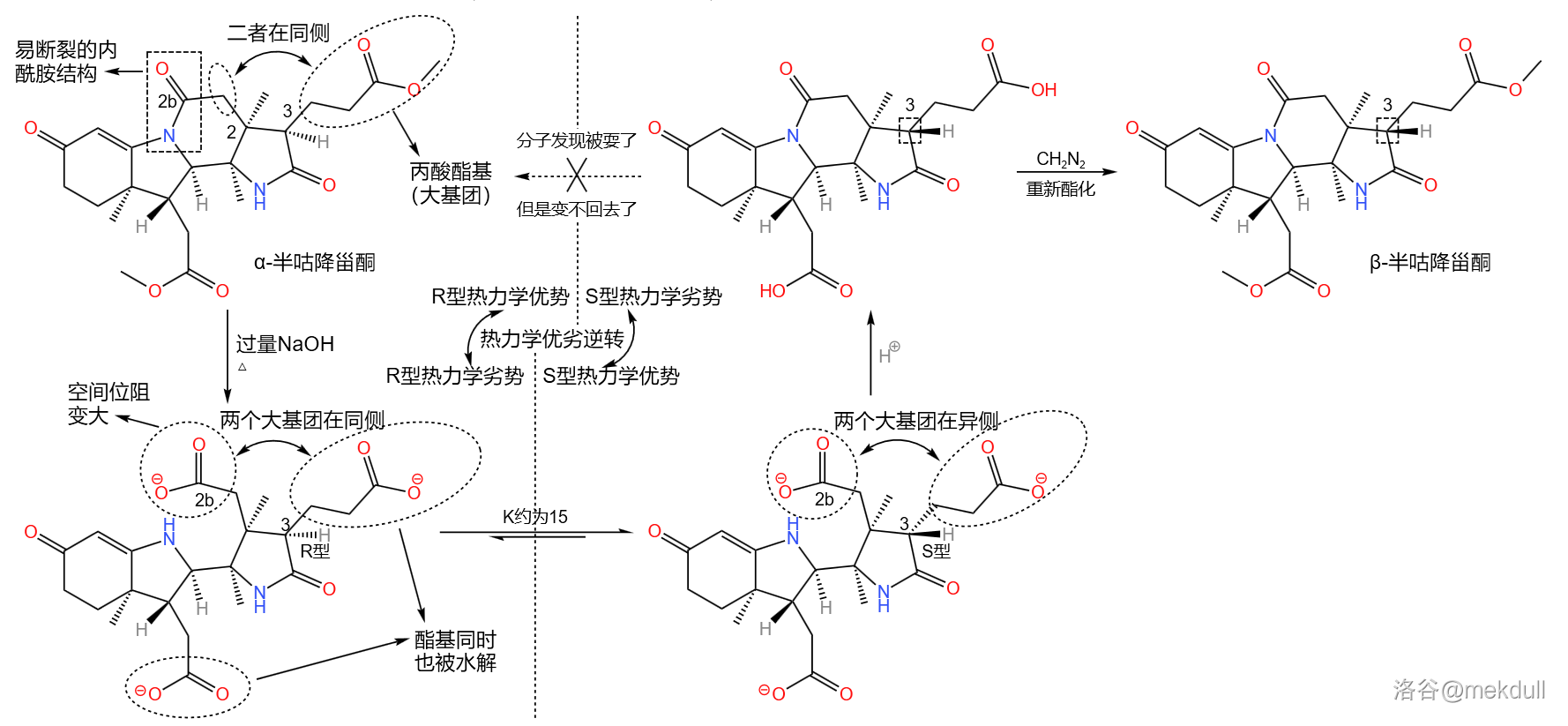
虽然构型翻转回来的只占了一小部分,但这给了课题组解决问题的希望。他们很快又发现,$2b$ 号位上的内酰胺结构可以在强碱的作用下断裂。于是,一个精妙的翻转构型的方法出现了。
观察 $2$ 号碳原子,你会发现它左侧的 $2b$ 号碳原子因为内酰胺结构而被“锁”在了一个固定的位置,连带着附近的 $2a$ 号碳原子也无法自由移动。这就相当于,$2$ 号碳原子的左侧是一个“动不了”的基团,所以空间位阻自然也比较小。而一旦我们用强碱把内酰胺环打开,那么这个基团就会重新“获得自由”,产生巨大的空间位阻。
那这又有什么用呢?前面提到,$3$ 号碳原子是特殊的,在合适条件(比如酸性或碱性条件)下,它的构型可以通过互变异构翻转。我们观察到,在 $\alpha-$半咕降甾酮中,$3$ 号碳原子右侧的丙酸酯基与 $2$ 号碳原子左侧的基团是位于同侧的。原来在被“锁住”时,由于空间位阻小,这种构型在热力学上是有优势的,但现在那个被锁住的已经“解放”了,空间位阻急剧增大,这种构型就变成了两个大基团贴在一起,热力学上的优劣就此逆转。于是,$3$ 号碳原子的构型翻转,从 $R$ 型变成了 $S$ 型。最后,我们只需要重新加酸至中性,分子就会重新环化——此时就算分子发现自己被耍了,构型也变不回去了,因为碱性条件已经丧失。就这样,我们成功把 $\alpha-$半咕降甾酮变成了需要的 $\beta-$半咕降甾酮:
接下来,在酸性条件下,用甲醇和苯硫醇($\ce{PhSH}$)的混合物处理 $\beta-$半咕降甾酮。由于活性差别,二者会分别进攻 $2b$ 号位和 $17c$ 号位上的羰基。最终,六元内酰胺环断开,$17c$ 号位上形成硫醚结构。
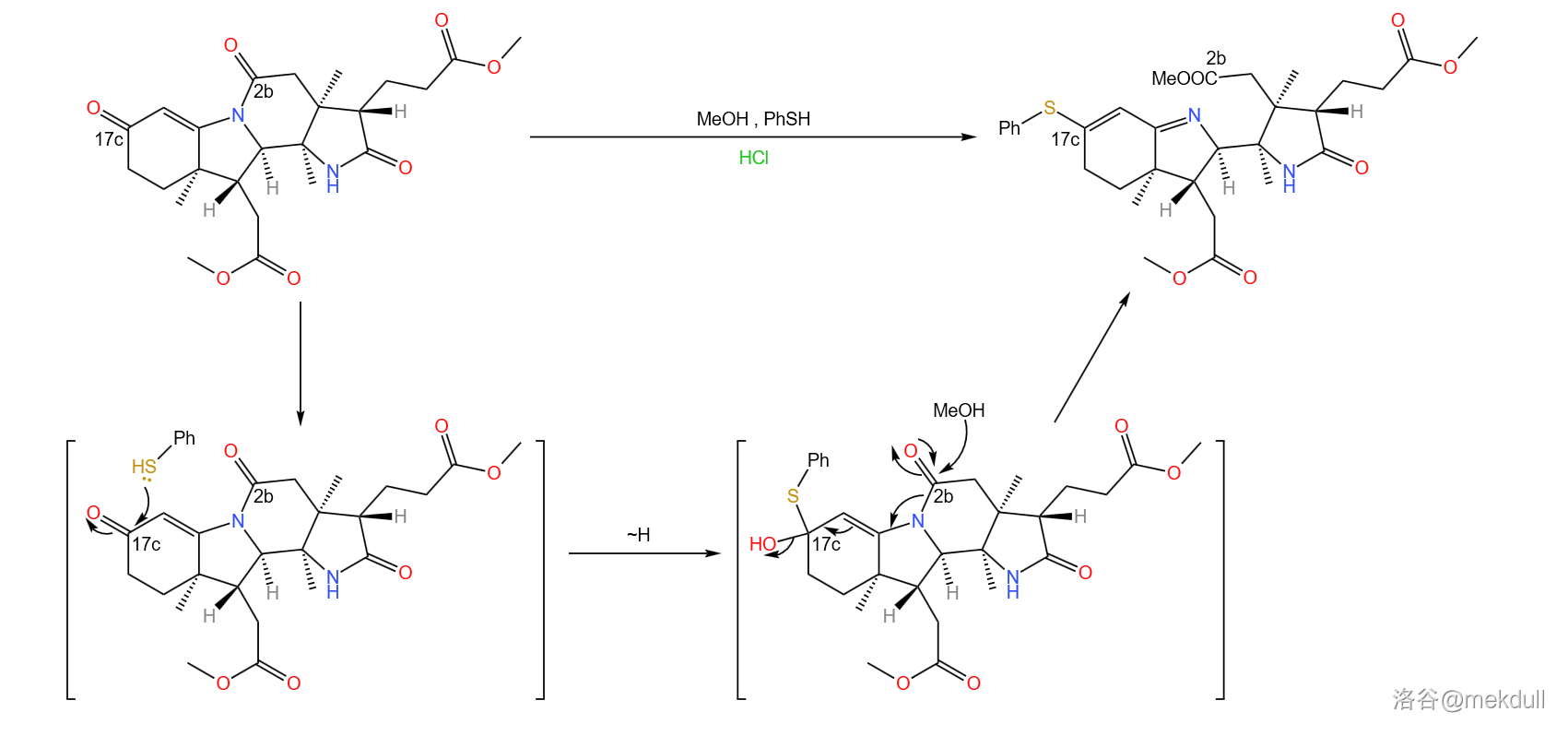
现在,我们手上的物质到底对应咕啉环的哪个部位已经可以看得很清楚了。如果你觉得不够清楚,那我们就把它旋转一下:
随后,在低温($-90^{\circ}C$)的甲醇中用臭氧处理该物质,双键断裂,并在 $17c$ 号位上生成 $\ce{-COSR}$ 的结构。接下来,用液氨处理之,$17c$ 号位上形成末端酰胺结构:
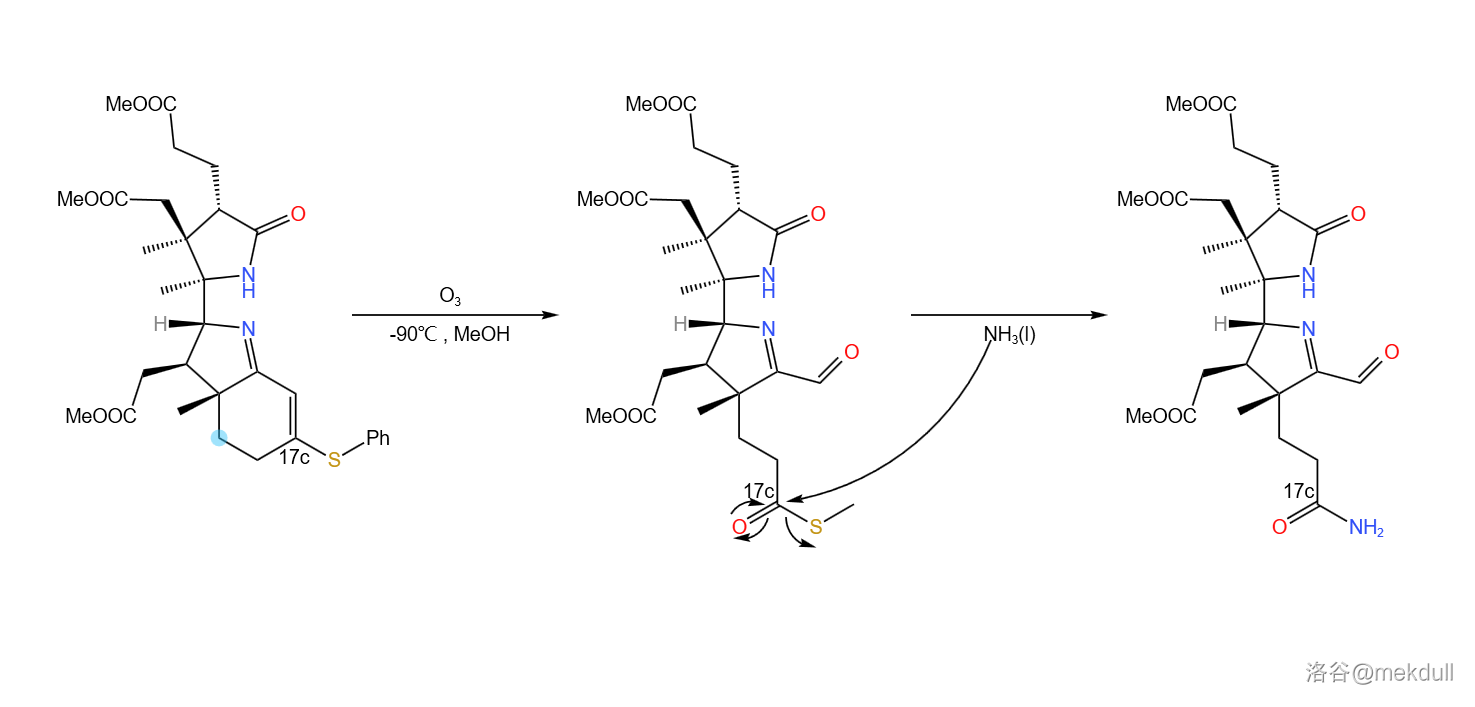
(注:其实 $\ce{-COSH}$ 这个基团是有一个专有名的,读作you基,$\ce{-COSR}$ 则名为you酯基,但我死活打不出来这个you字,还请各位指教;下文中我会把 $\ce{-COSR}$ 称为“硫酯”,而把 $\ce{-CSOR}$ 称为“硫羰酯”,请注意区分)
最后一步,在还原剂硼氢化钠($\ce{NaBH4}$)的作用下,$15$ 号位上的醛被还原为醇;加入过量的甲磺酸酐($\ce{MsOMs}$),将醇甲磺酰化的同时为 $17c$ 号位上的末端酰胺脱水,得到一个氰基($\ce{-CN}$);最后加入溴化锂,左半部分“$cyanobromide$”的全合成就大功告成了:
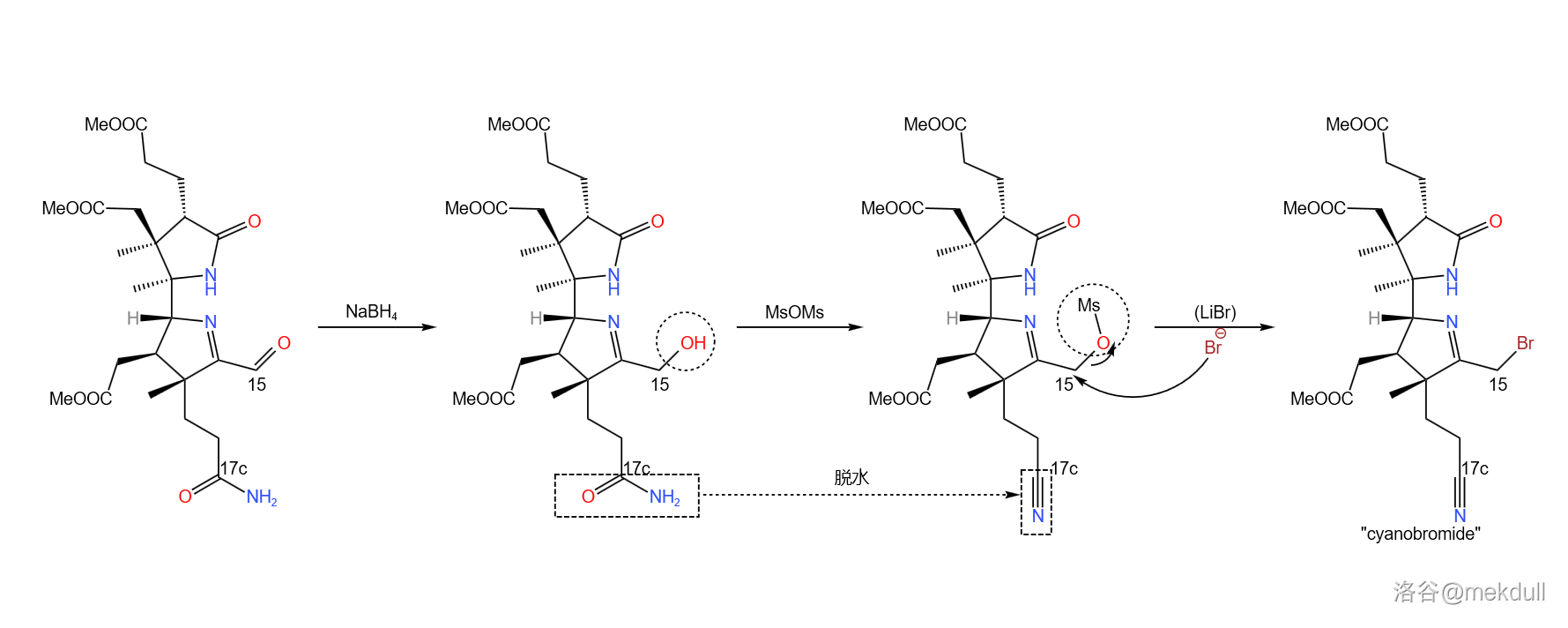 然而,这也只是一半而已。接下来,舞台交给 $Eschenmoser$ 的课题组,他们将要带我们去看看,分子的右半部分(“$thiodextrolin$”)是怎么合成出来的。
然而,这也只是一半而已。接下来,舞台交给 $Eschenmoser$ 的课题组,他们将要带我们去看看,分子的右半部分(“$thiodextrolin$”)是怎么合成出来的。
3.2 右半部分的合成
相比于 $Woodward$ 课题组的工作,$Eschenmoser$ 课题组设计的合成路线更为简短,但同样精妙。
右半部分的合成起始于三种结构简单、产量巨大的重要化工原料:乙醛酸($\ce{HOCCOOH}$)、丁酮($\ce{CH3COCH2CH3}$)和丁二烯($\ce{CH2=CHCH=CH2}$)。前两者在酸催化下发生交叉羟醛缩合反应,得到一种烯基酮酸;随后,在四氯化锡($\ce{SnCl4}$,作为路易斯酸)的催化下,该物质与丁二烯发生著名的 $Diers-Alder$ 反应,得到一种环己烯的衍生物:
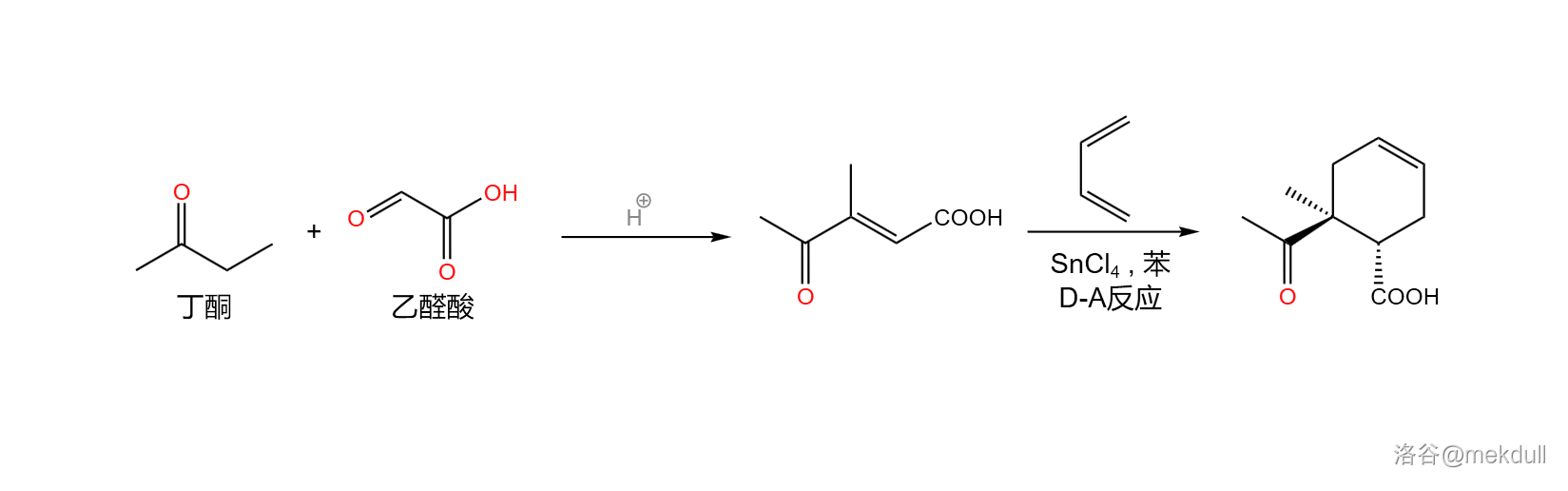
$Diers-Alder$ 反应(又名双烯加成反应)在有机合成中非常重要,其机理在前几篇文章中已有提及,而在这篇文章的后记中,我们还会简要讨论全合成工作为这个反应带来的发展。
接下来,用过量的 $Jones$ 试剂(三氧化铬($\ce{CrO3}$)溶于浓硫酸)处理该物质,环状烯烃结构的双键随即断裂(此处反应条件非常强烈,一般来说 $Jones$ 试剂不会氧化双键),生成一种带有三个羧基的中间体;该中间体随后自发环化,形成双内酯结构,产率 $75\%$:
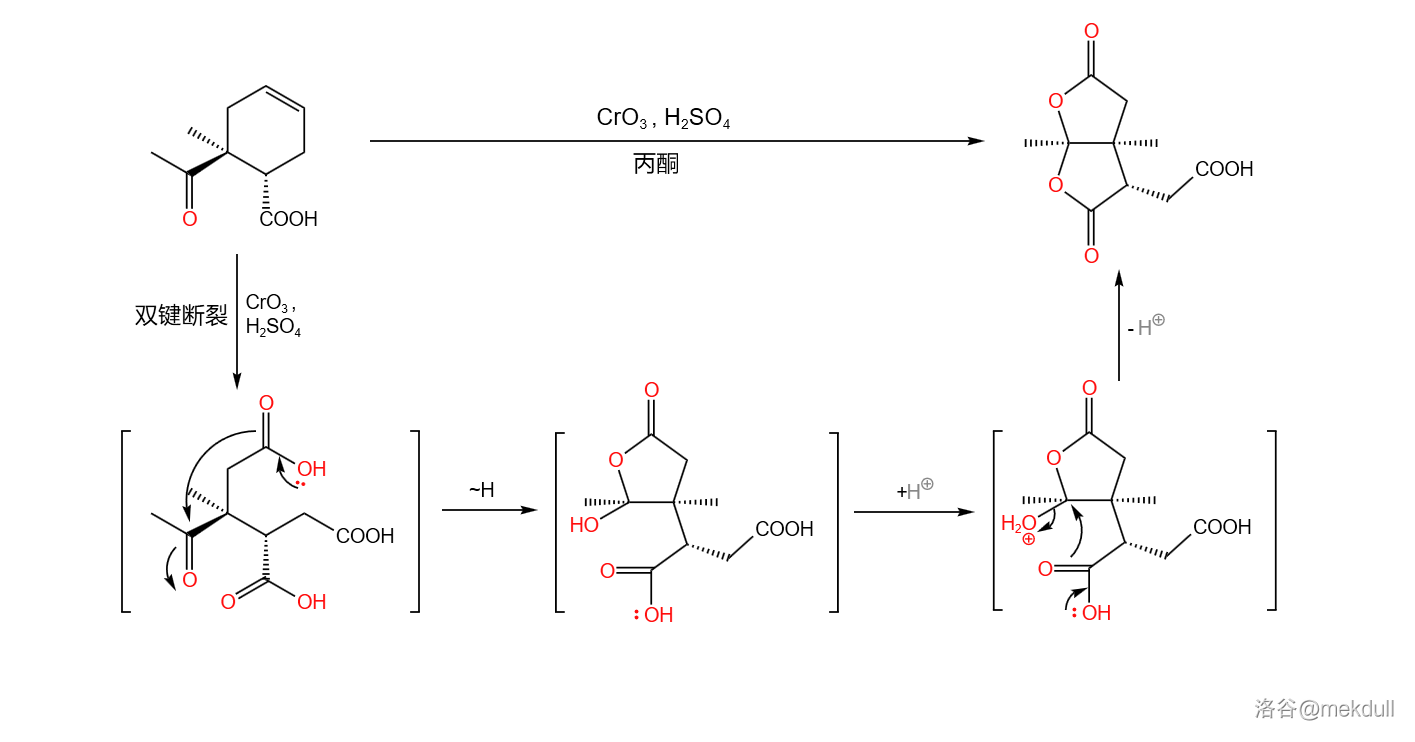
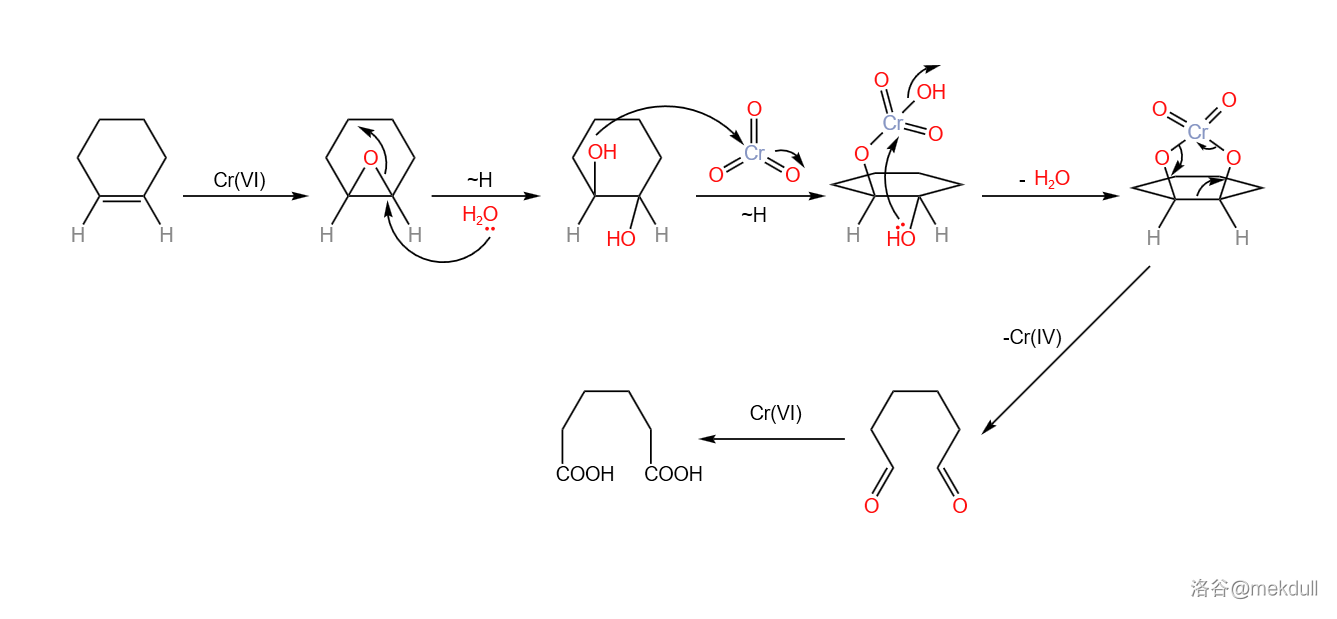
关于 $\ce{Cr(VI)}$ 氧化剂断裂双键的机理,一般认为是先生成环氧化合物,随后生成邻二醇,最后在过量氧化剂的作用下,碳碳键断裂,得到二酸产物:
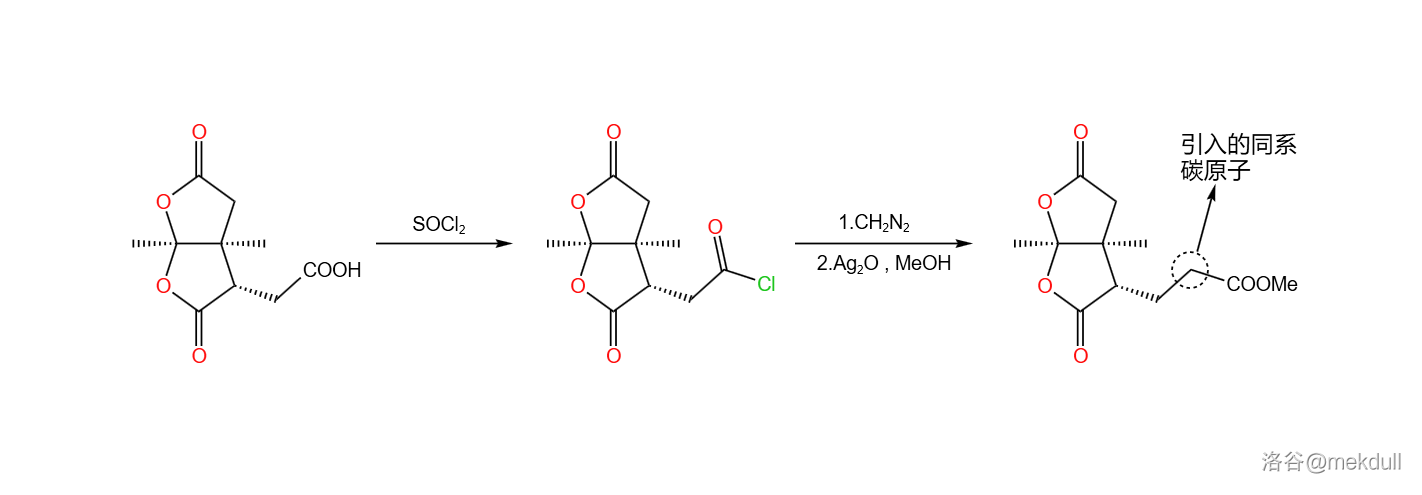
言归正传。下一步要用到的是 $Arndt-Eistret$ 同系增碳反应,顾名思义,这个反应可以为羧基增加一个同系碳原子。在 $\ce{SOCl2}$ 作用下,分子内的羧基首先变成酰氯;加入 $\ce{CH2N2}$,在亲核取代之后,同系碳原子随即被引入;在 $\ce{Ag(I)}$ 的催化下,分子放出 $\ce{N2}$ 形成卡宾酮中间体,随后发生重排反应,最终在 $\ce{MeOH}$ 的作用下变成酯,三步反应总产率 $69\%$:
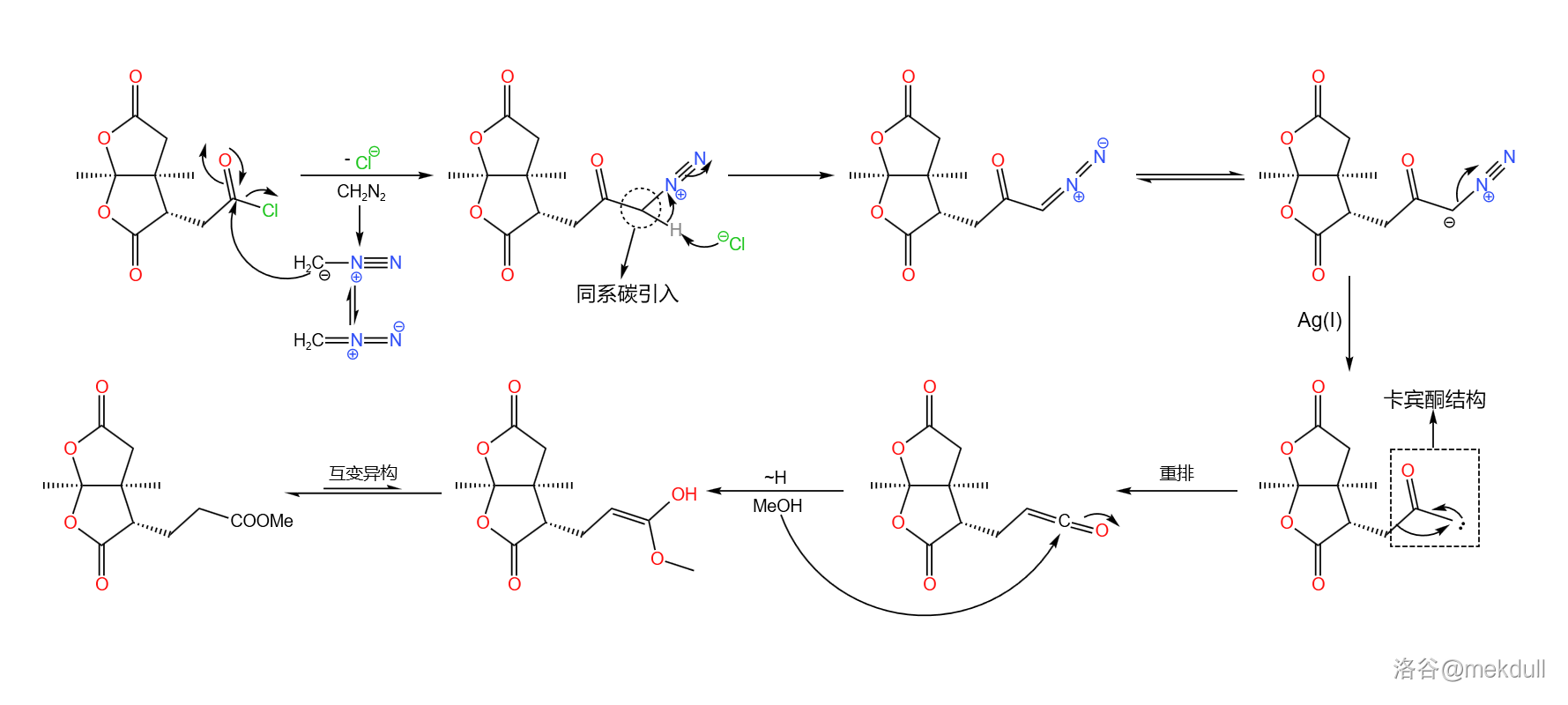
随后,在甲醇中用定量的 $\ce{NH3}$ 处理该物质,可以将其中一个内酯基团转化为内酰胺。由于特殊的双内酯结构,该物质的羰基具有更强的亲电性,所以在室温下反应就可以快速进行:
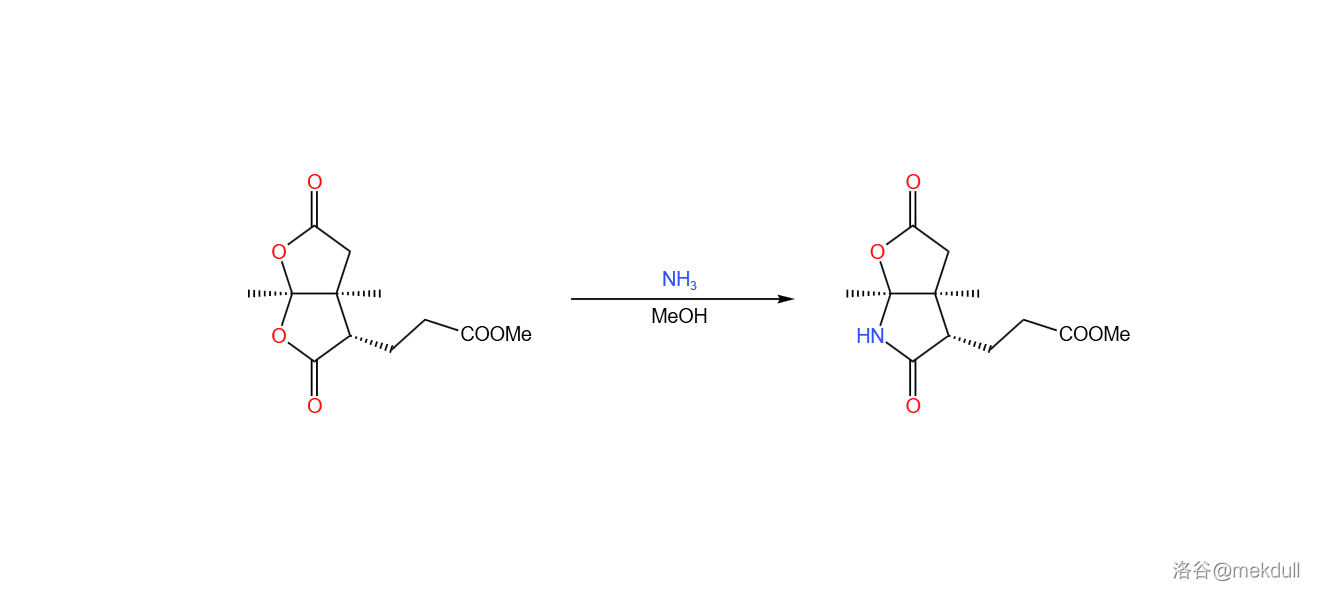
反应产率约为 $55\%$(其余的 $45\%$ 大多都是另一个内酯基团被变成了内酰胺)。可见,这个反应的选择性其实并不显著。
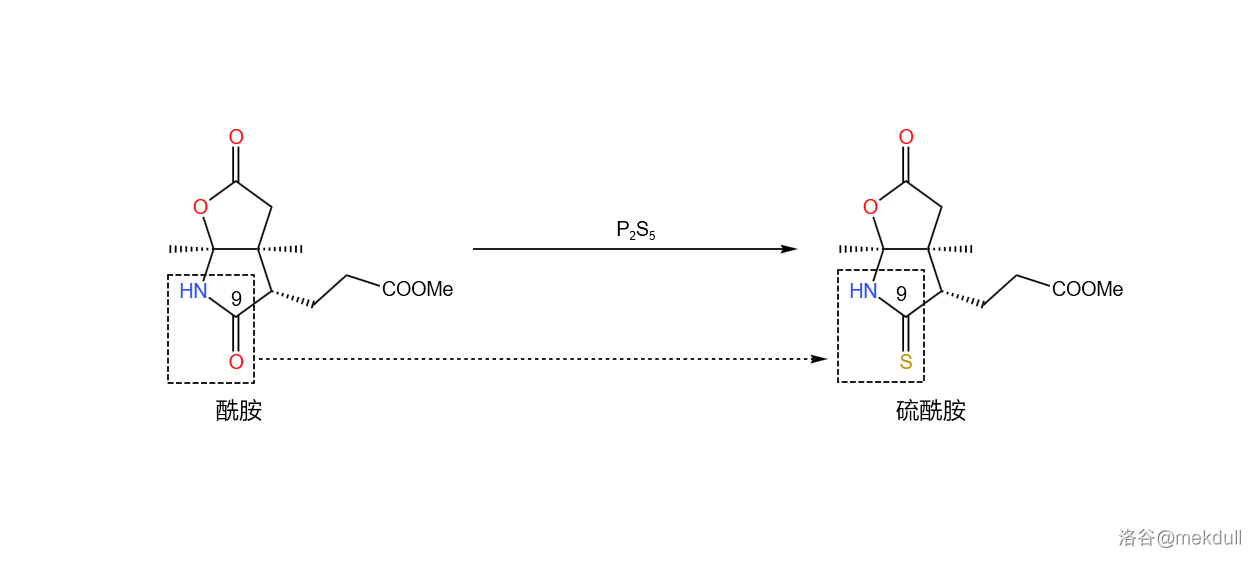
但不管怎样,$55\%$ 的产率也算是能接受了。现在让我们把产物分成两份,并对它们进行不同的处理。第一份的处理很简单,在 $\ce{THF}$ 中,用一定量的五硫化二磷($\ce{P2S5}$)进行硫化,$9$ 号位的内酰胺结构被转化为硫内酰胺,而分子中的其他酯基则不受影响:
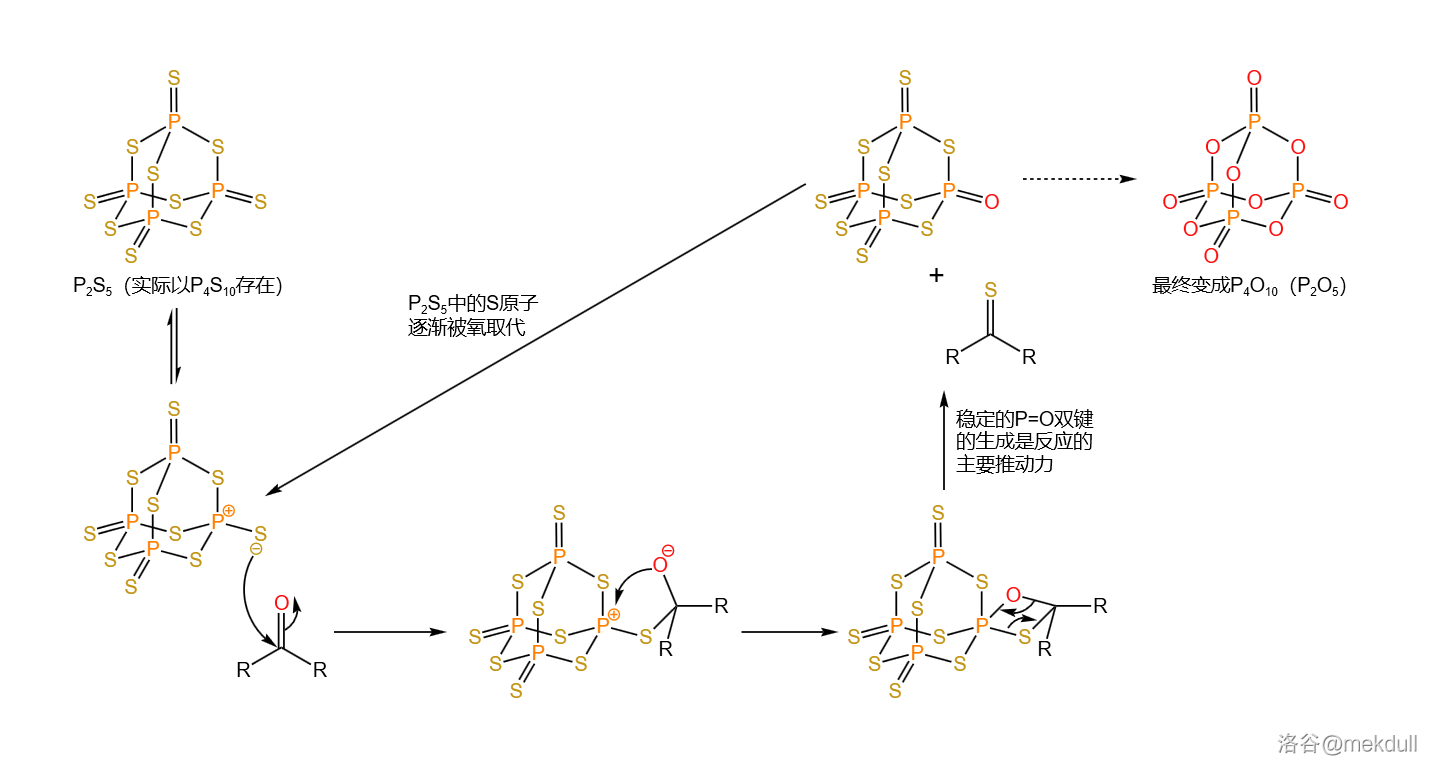
五硫化二磷($\ce{P2S5}$)的分子结构与所谓五氧化二磷($\ce{P2O5}$)类似,实际上是以 $\ce{P4S10}$ 形式存在的。作为一种重要的化工原料,它在有机合成中经常被用于将羰基转化为硫羰基。后来,人们又以它为基础制得了威力更强的版本,也就是著名的劳森试剂($Lawesson’s~Reagent$)。$\ce{P2S5}$ 硫化的机理如下,有点类似于 $Wittig$ 反应的机理:
扯得有点远,让我们来看看第二份是怎么处理的。在乙醚($\ce{Et2O}$)中,用 $\ce{NaOMe}$ 裂解内酯环,随即消去 $1$ 分子水生成 $10,11$ 号位上的双键;为了防止需要的酯基被水解,反应体系中还添加有一定量的 $\ce{CH2N2}$。最终产率约为 $\ce{92\%}$:
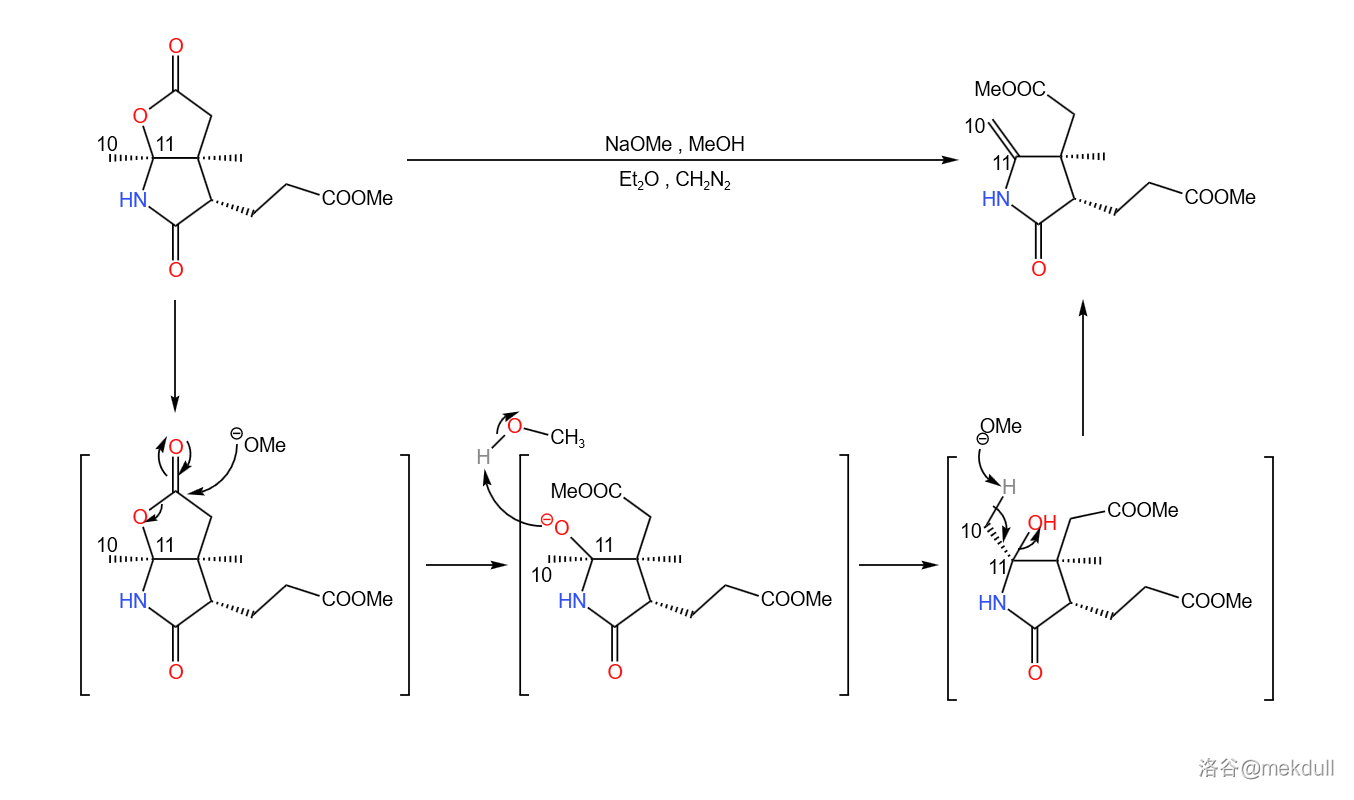
接下来,在三氟乙酸($\ce{CF3COOH}$,缩写 $\ce{TFA}$)的作用下,该物质与 $\ce{H2S}$ 加成(与加 $\ce{H2O}$ 的机理类似)。随后,硫原子进攻附近的酯基,形成五元环状硫酯($\ce{-CSOR}$)结构:
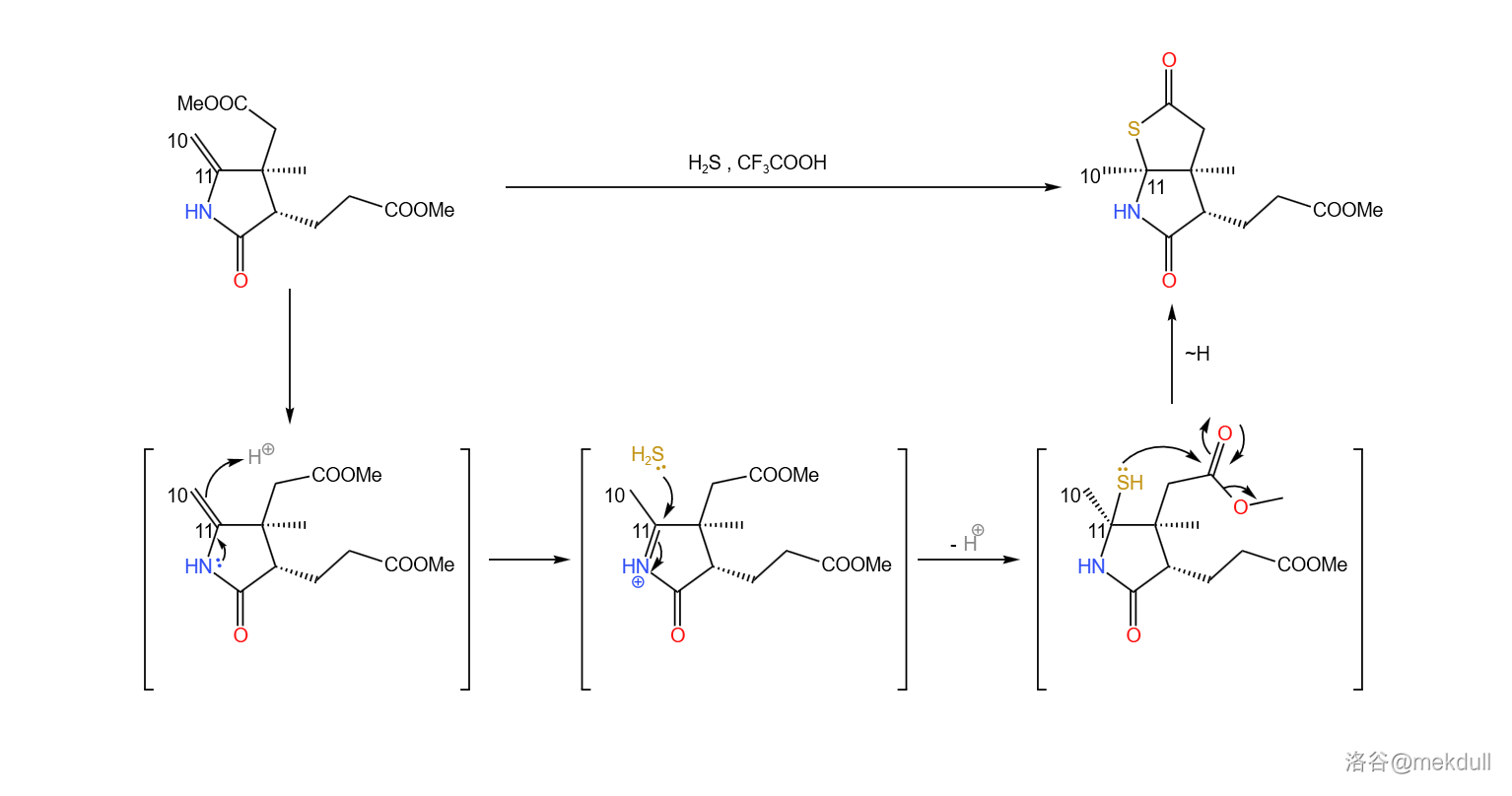
之所以要大费周章把它搞成硫酯,是因为在威尔金森催化剂($\ce{(Ph3P)3RhCl}$)和氰化钠($\ce{NaCN}$)的作用下,硫酯容易发生脱硫化羰反应,从而把那个不需要的碳原子去掉;随后再用 $\ce{Ag+}$ 处理,得到 $10,11$ 号位上的双键。这个反应同时也会产生一种有三元环结构的副产物,因此产率并不是很高:
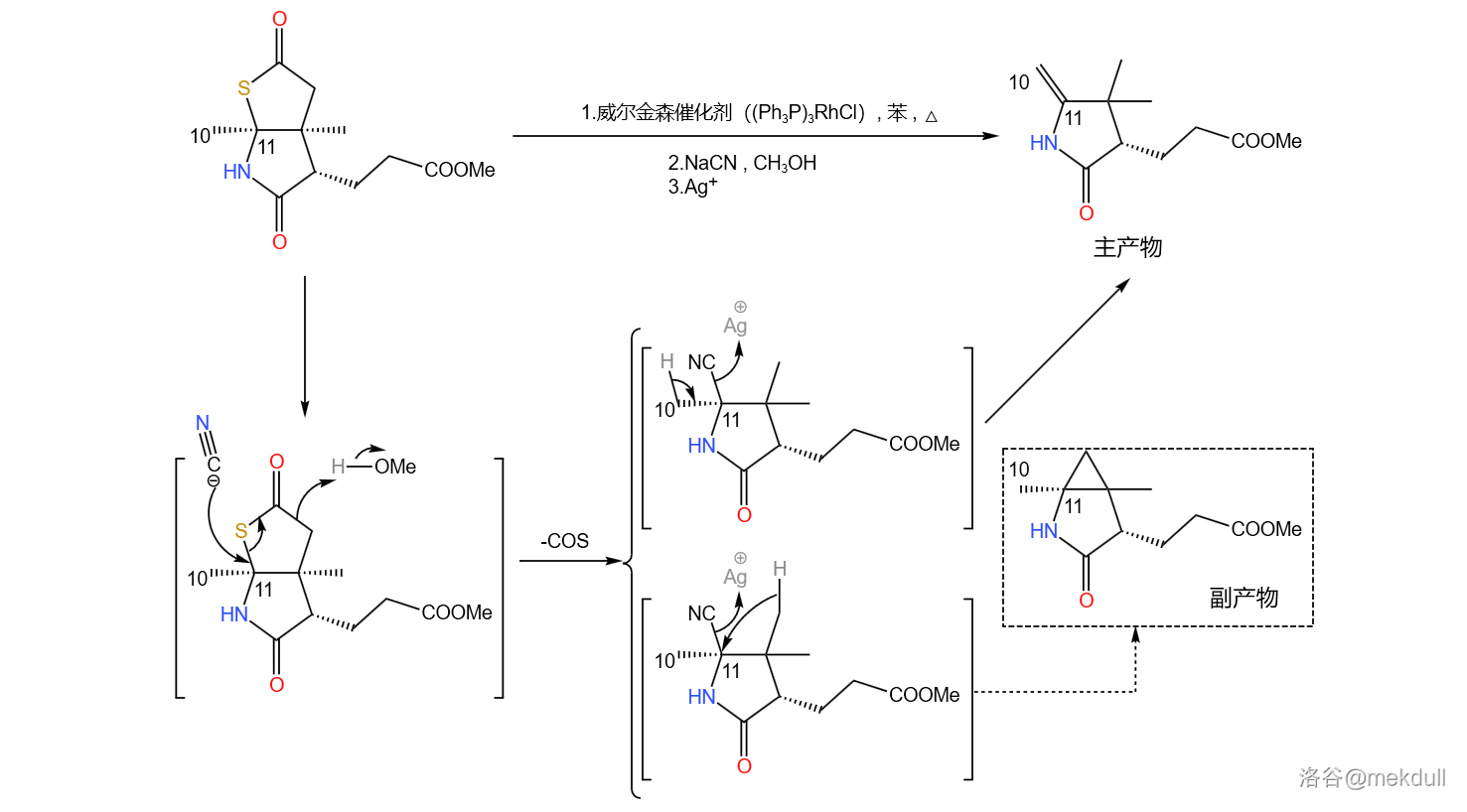
(注:由于涉及过渡金属催化,机理相当复杂,所以此处展示的机理是简化过后的。想要进一步了解的读者可以自行查阅相关文献。)
好,现在两份都已经完成了各自的处理,是时候把它们结合起来了。为了这一步,$Eschenmoser$ 开创了一个全新的反应,也就是如今已被我们所熟知的 $Eschenmoser$ 偶联反应。
$Eschenmoser$ 偶联反应需要一个硫酰胺和一个烯胺。在过氧化苯甲酰($\ce{PhCOOOCOPh}$)的作用下,两分子硫酰胺首先发生氧化自偶联;随后,烯胺进攻自偶联产物,二硫键断裂得到硫桥结构,又自发异构化为环硫化合物;最后在 $\ce{P(III)}$ 化合物(此处使用三乙氧磷,$\ce{P(OEt)3}$)作用下,硫原子被拔去,完成整个偶联反应:
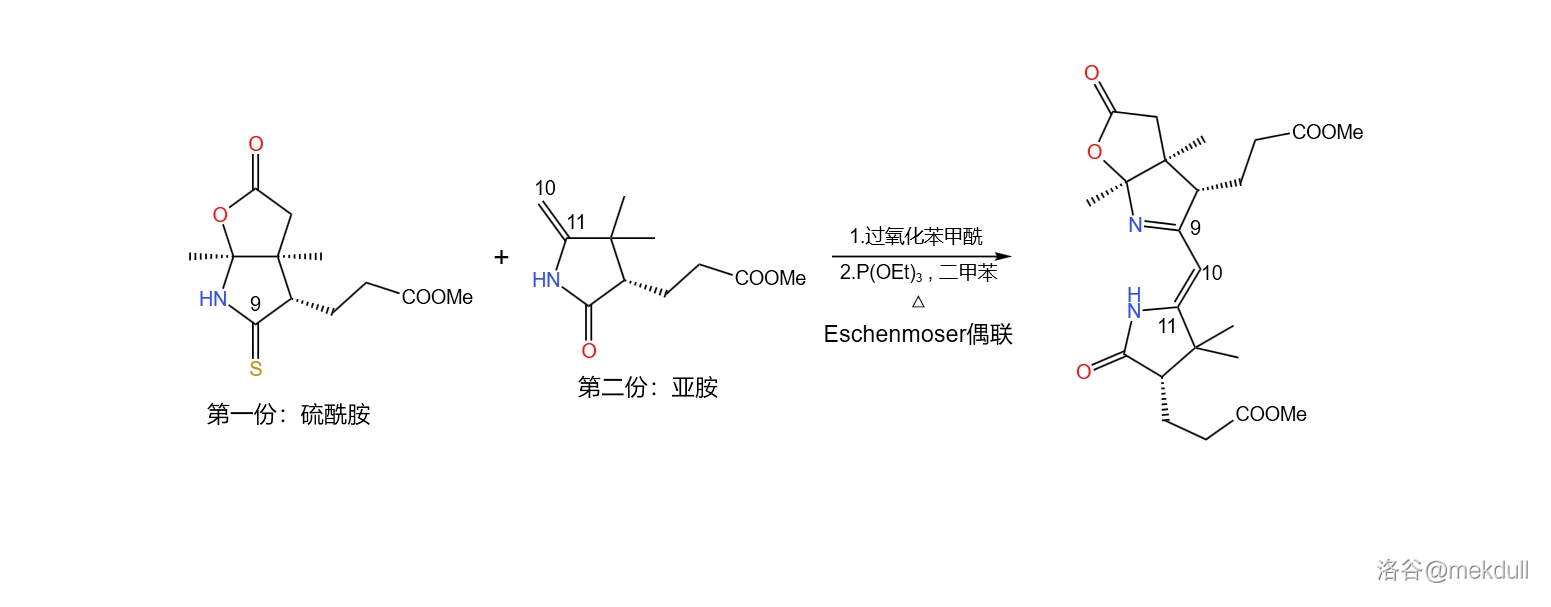
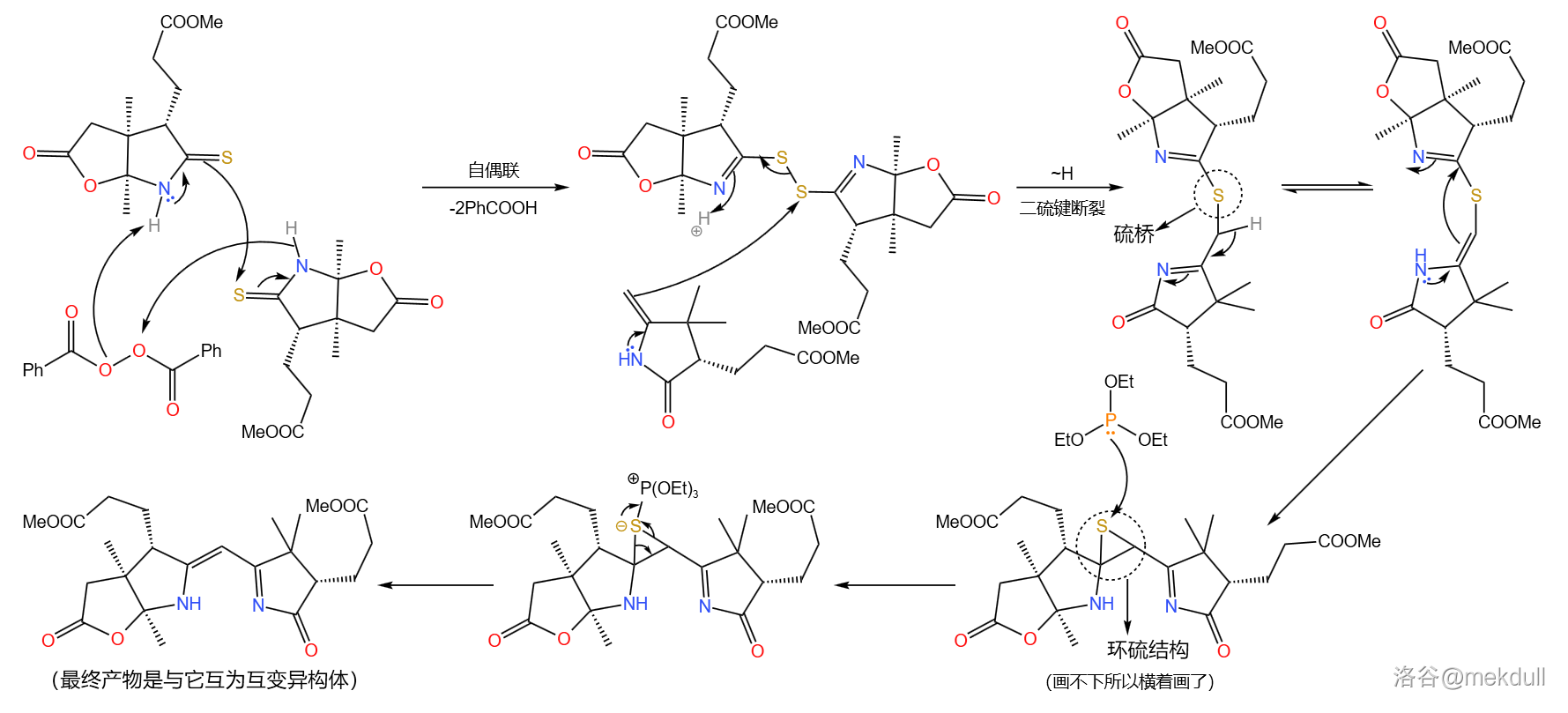
必须说,这个反应设计得非常巧妙,但它的通用性并不好,因为底物之一的烯胺与亚胺是互变异构体,且一般平衡偏向后者。因此,有人对这个反应进行了改进,而稍后我们就会看到这种变异的 $Eschenmoser$ 偶联反应。
偶联反应之后,距离完成只差最后一步了——将 $14$ 号位的酰胺结构硫化。这一次,分子内多出了许多结构,为了保证它们不被破坏,课题组在多次实验后选择了在甲基汞离子($\ce{MeHg+}$)催化下用 $\ce{H2S}$ 直接硫化的方案,并最终以高产率得到了目标产物:
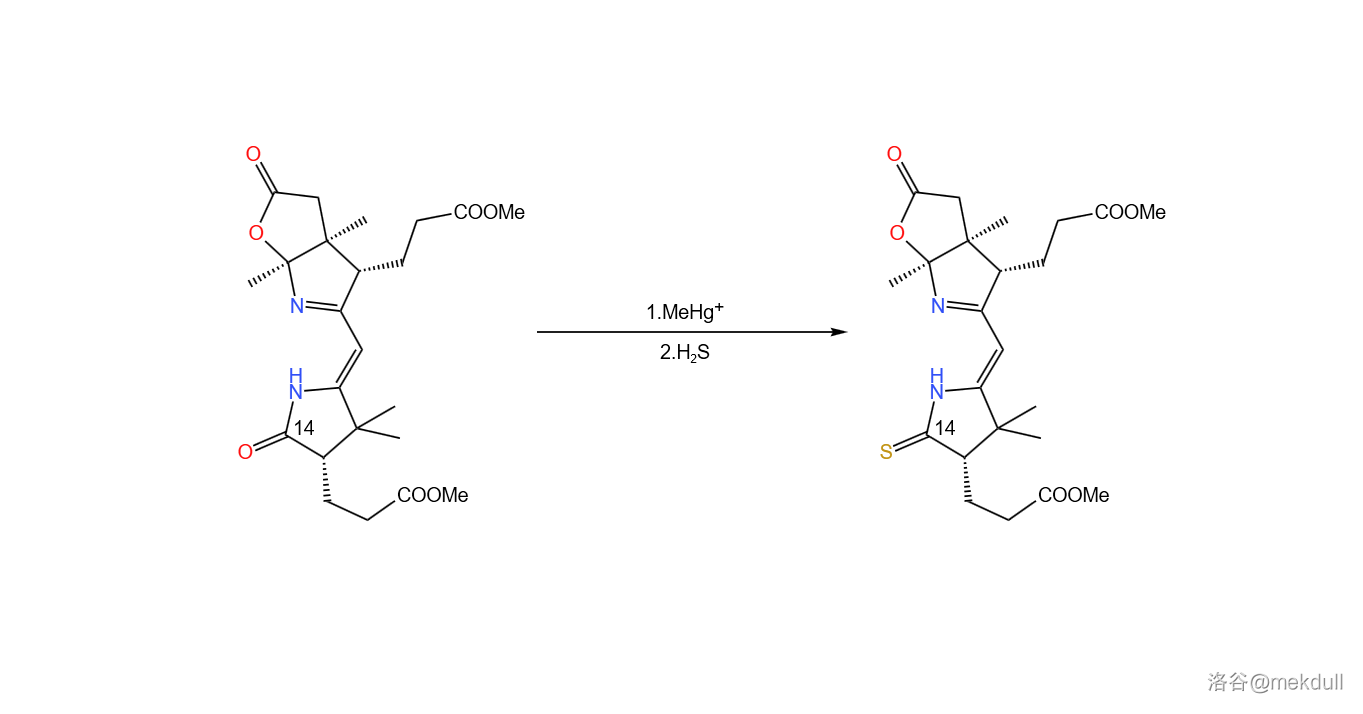
就这样,右半部分“$thiodextrolin$”的全合成也宣告成功。接下来,就该把它们拼起来了。
3.3 咕啉环的拼接与修饰
这一部分的工作是两个课题组各自完成的,但篇幅所限,我们主要来讲一讲 $Woodward$ 课题组设计的合成路线。(事实上,两条路线的大体思路是基本一致的)
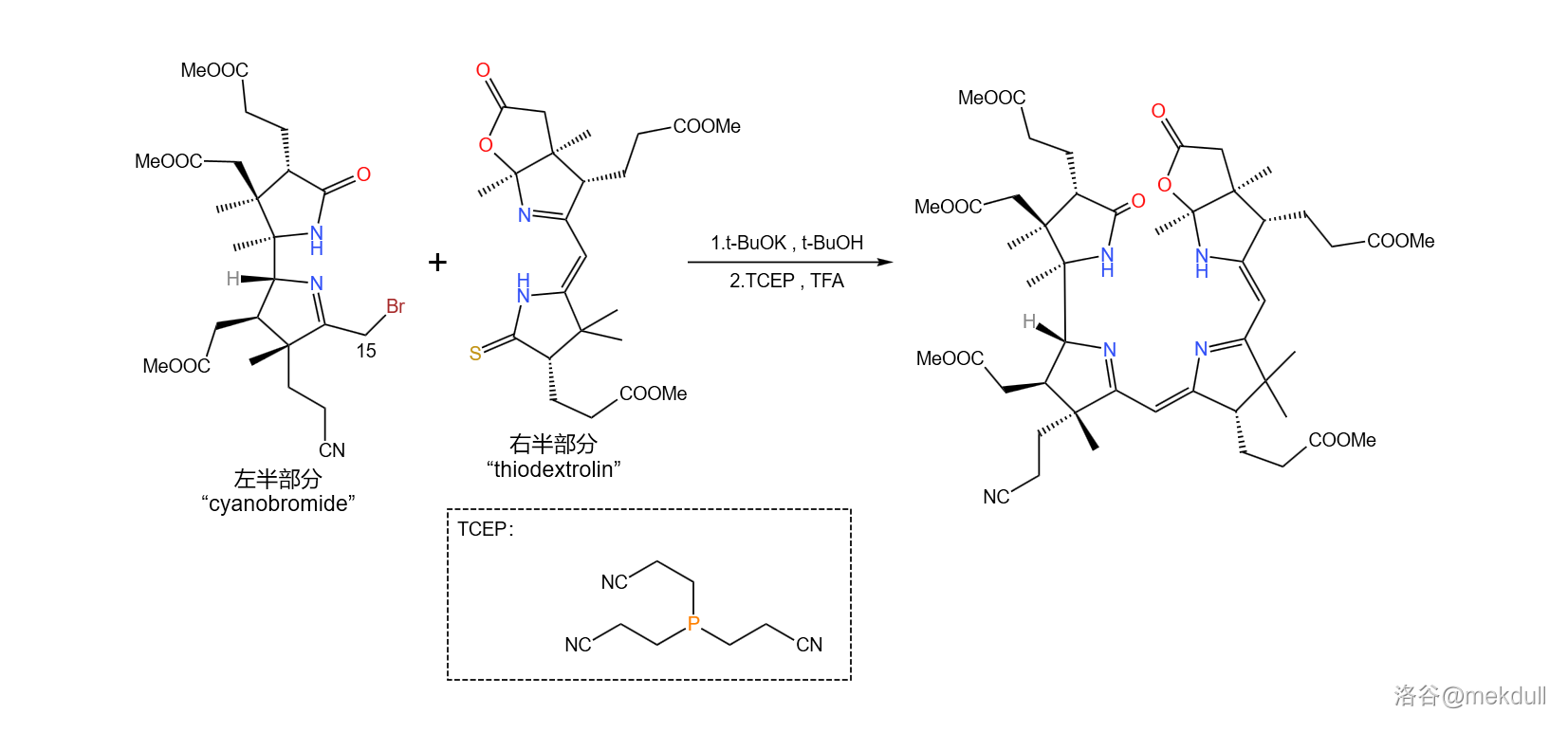
拿到了左右两半后,首要的任务当然是将它们先拼在一起。$Woodward$ 果断采用了 $Eschenmoser$ 偶联的一种变异反应。这种变异将其中一种底物从烯胺变成了活泼卤代烃,从而使反应的通用性大大增强;利用它,就能轻松地在 $15$ 号位上完成偶联:

接着,在 $4-$甲基吡啶催化下用一定量的 $\ce{P2S5}$ 处理之,$4$ 号位的内酰胺和 $7b$ 号位上活泼的内酯结构被硫化,分子中其他普通的酯基不受影响;随后加入 $Meerwin$ 盐对硫羰基进行甲基化:
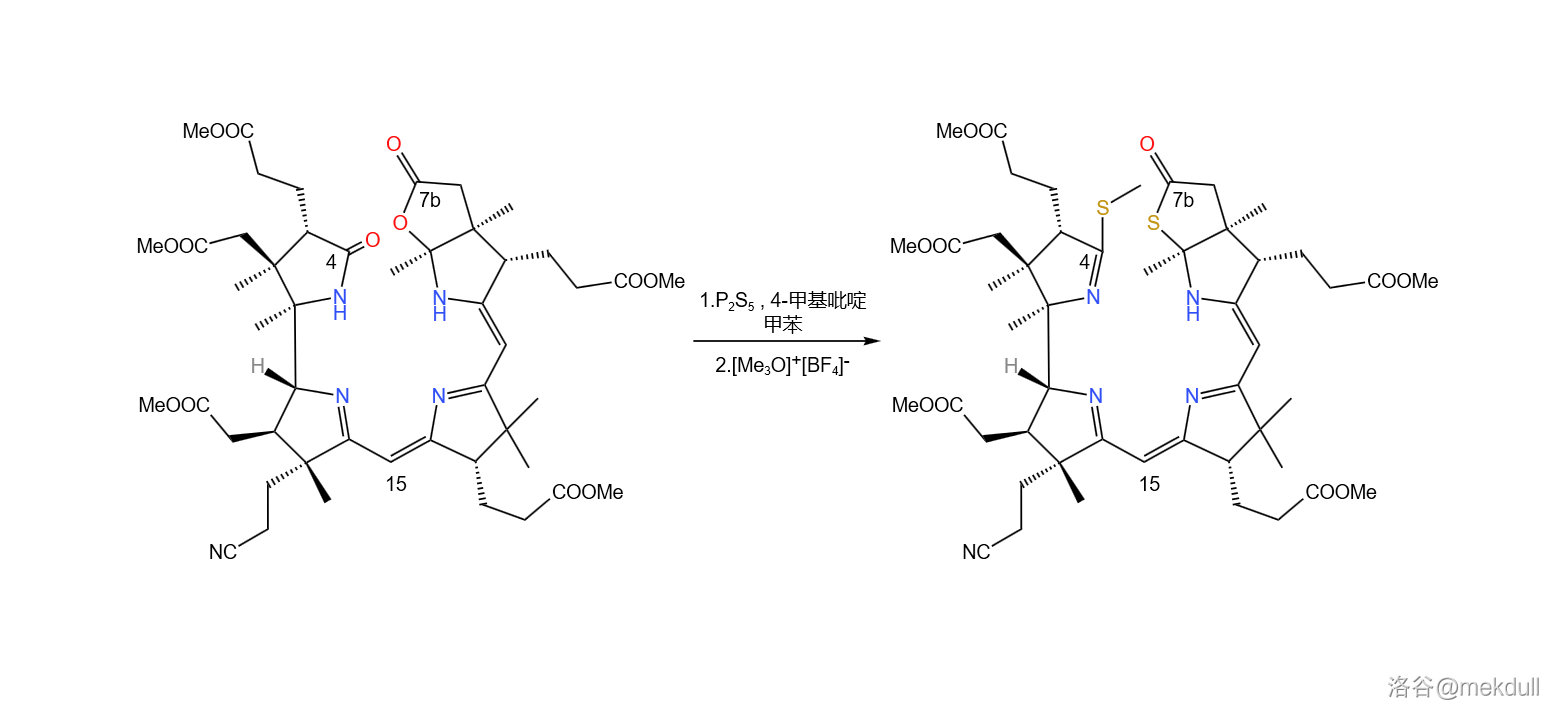
补充一句,$\ce{P2S5}$ 在硫化酯基时一般先会生成硫羰酯,也就是 $\ce{-CSOR}$ 结构,但在碱性试剂作用下,硫羰酯会变成通常更稳定的硫酯($\ce{-COSR}$)结构。对 $7b$ 号位内酯结构的硫化就大致经历了这一历程。
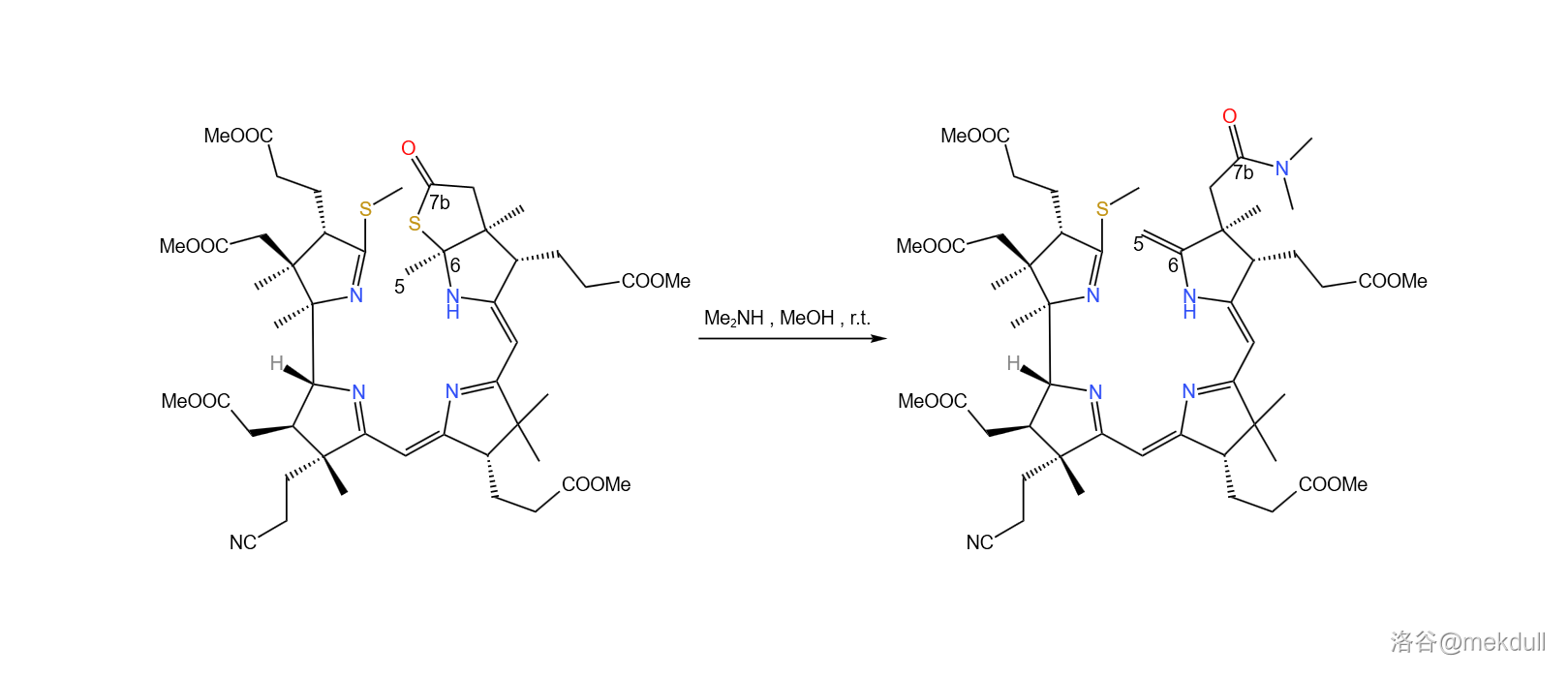
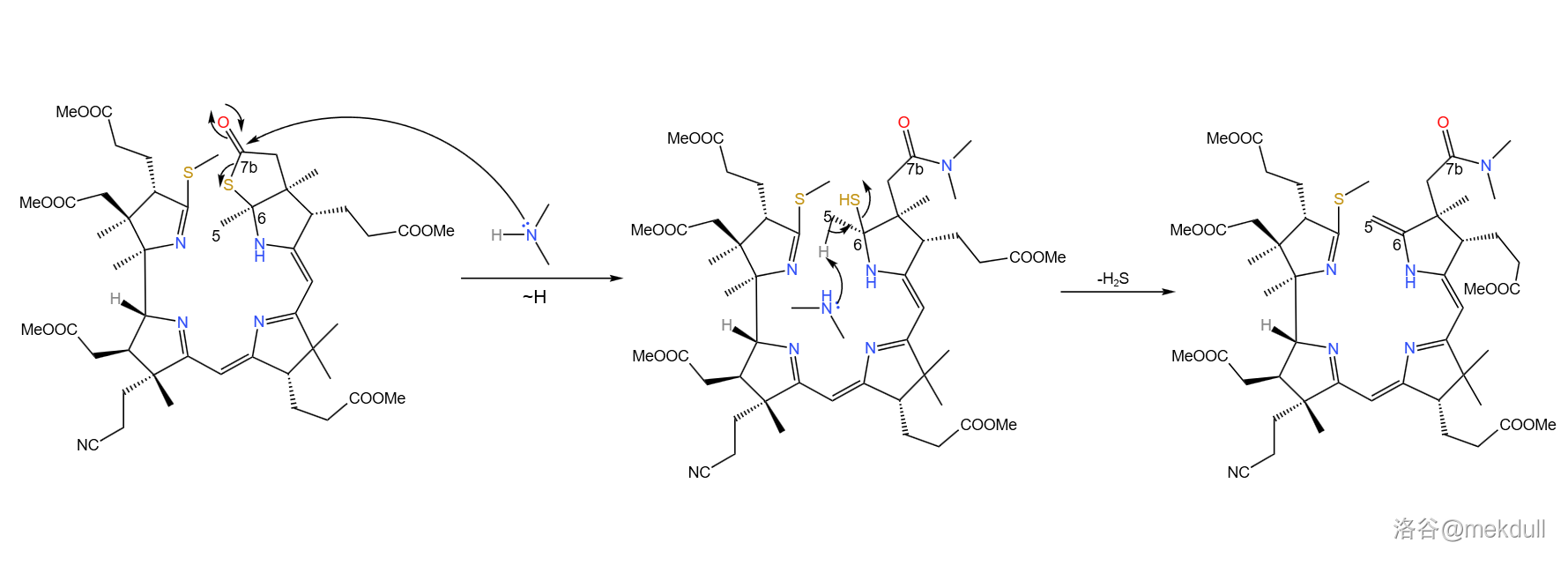
下一步,使用二甲胺($\ce{Me2NH}$)打开硫内酯环,得到 $7b$ 号位的酰胺结构,同时在 $5,6$ 号位上发生消去反应,生成双键。这个反应看似简单,但课题组却花费了不少时间去修正反应条件,才使反应的产率足够令人满意:
随后,课题组打算引入中心配位的 $\ce{Co}$ 原子。一开始,课题组并没有预料到这一步的困难,但他们马上就会知道了:$\ce{Co}$ 是一种极其有效的催化剂,可以迅速催化该物质分解,让前面的一切努力都变成白忙活。有人会问:为什么一定要这么着急地把 $\ce{Co}$ 原子放进去呢?因为在下一步,我们将要进行 $4$ 号位和 $5$ 号为碳原子的偶联。还是由于空间位阻,此时 $4$ 号和 $5$ 号碳原子虽然在纸面上看起来很近,但实际上距离很远——这就导致分子内的偶联反应难以进行。下图是由计算机渲染得到的,大家可以直观感受一下两者的距离:
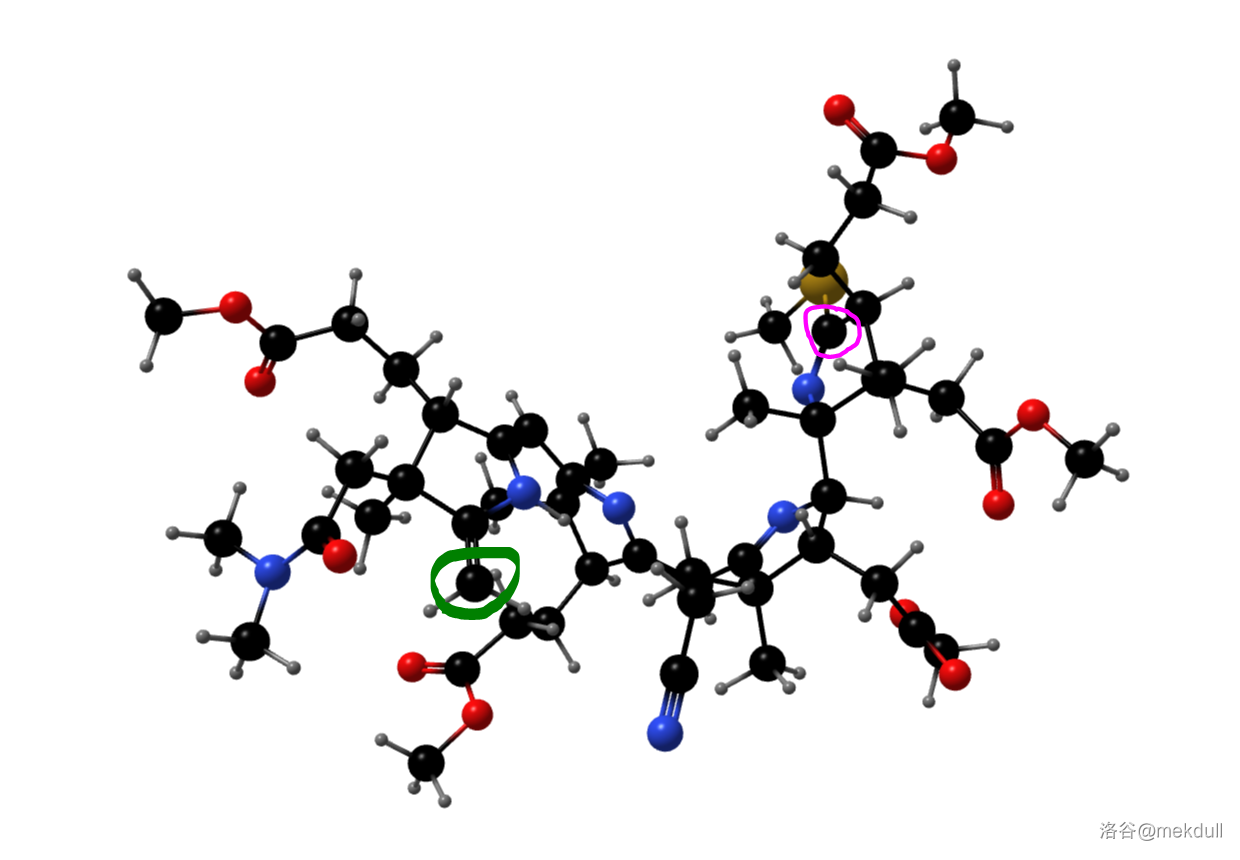
(上图:目前我们手中化合物的空间结构,绿色圈出的是 $5$ 号碳原子,粉色圈出的是 $4$ 号碳原子)
如果此时我们成功地将中心的 $\ce{Co}$ 原子塞进去,那么整个分子的空间结构都会因此改变。$4,5$ 两个碳原子会被强行“拉拢”,从而使得它们之间的偶联反应可以顺利进行。想到这里,$Woodward$ 下定决心要攻克这道难题。它一定是有解的!维生素B12就在那里,它的存在,就是问题有解的最好证明!带着这份信念,课题组花了整整一年的时间,尝试各种方法和反应条件,同时也尝试揭秘 $\ce{Co}$ 催化分解的机理。最终,他们找到了。
在最终的目标中,$\ce{Co}$ 原子的氧化数应为 $\ce{+III}$,但在几百次实验之后,课题组确信:正是 $\ce{Co(III)}$ 破坏了分子的结构。所以,最终的解法就是:先在 $\ce{THF}$ 中用无水氯化钴($\ce{CoCl2}$)处理,让 $\ce{Co(II)}$ 进入配位体系;随后加入 $\ce{KCN}$ 水溶液,利用 $\ce{CN-}$ 离子强大的配位能力把 $\ce{Co(II)}$“锁”在分子的中心;等到万事俱备了,再将 $\ce{Co(II)}$ 用空气氧化成 $\ce{Co(III)}$。此时,就算是 $\ce{Co(III)}$ 想搞破坏也不可能了:
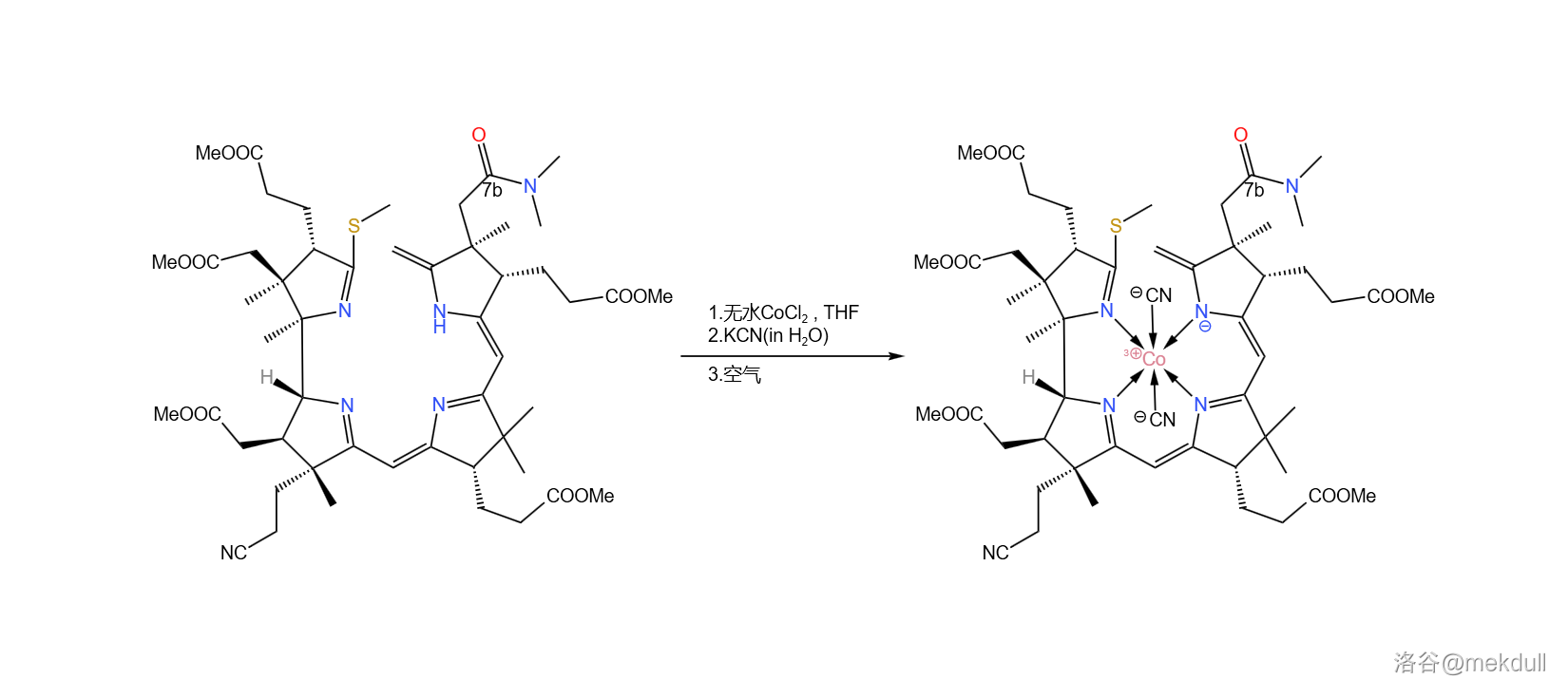
补充一句,此处的无水氯化钴也不是随便选择的。很早之前就有人发现,$\ce{Cl-}$ 和 $\ce{I-}$ 似乎对于钴络合物的形成具有催化作用,虽然这种作用的机理尚不明确。到了这里,如果不用无水卤化物的话,那么 $\ce{Co(II)}$ 会很难进入配位体系,也就没法进行下一步的反应了。
难题解决之后,后面的反应就如同预料一般顺利了。在 $\ce{DMAC}$(二甲基乙酰胺,$\ce{CH3CON(CH3)2}$)中用有机碱 $\ce{DBN}$ 进行催化,偶联反应就可以高产率地进行:
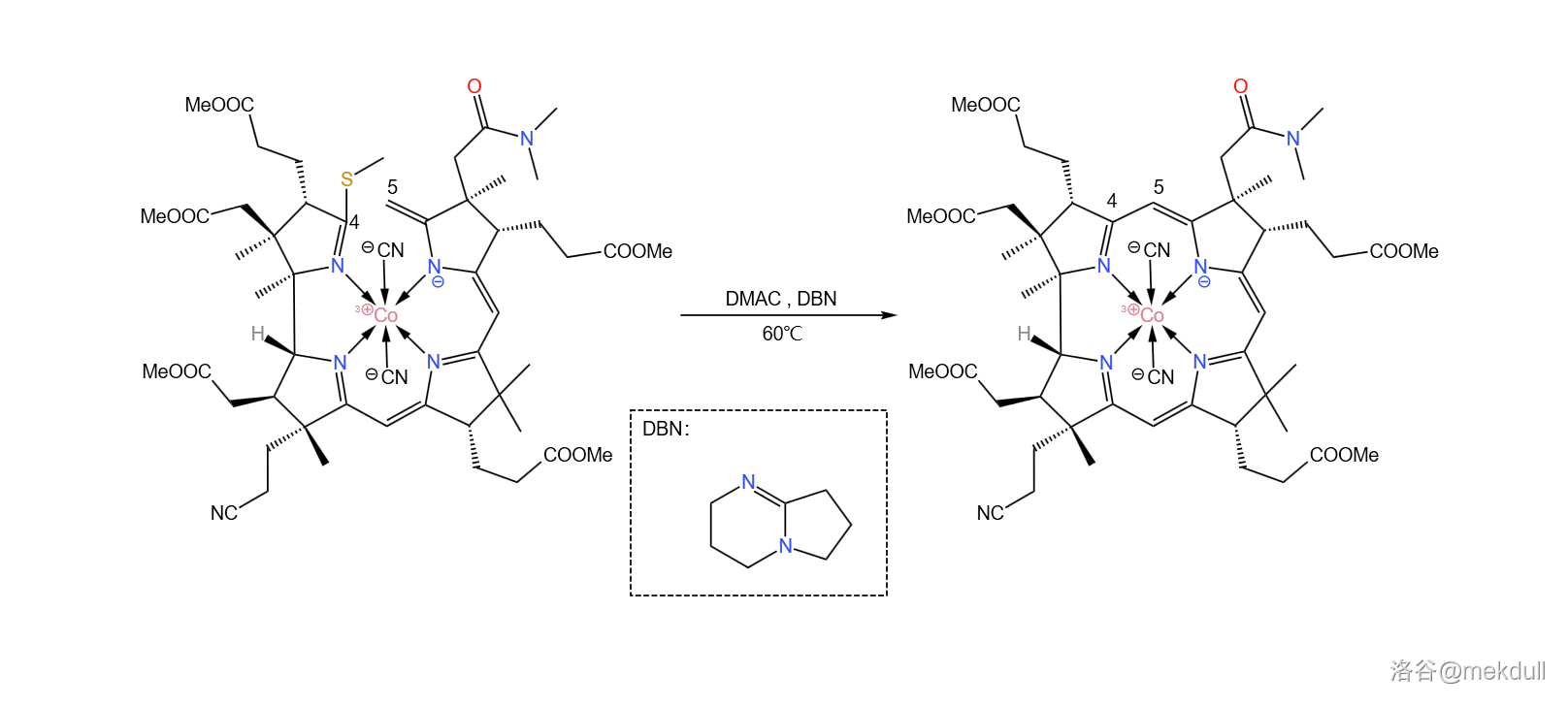
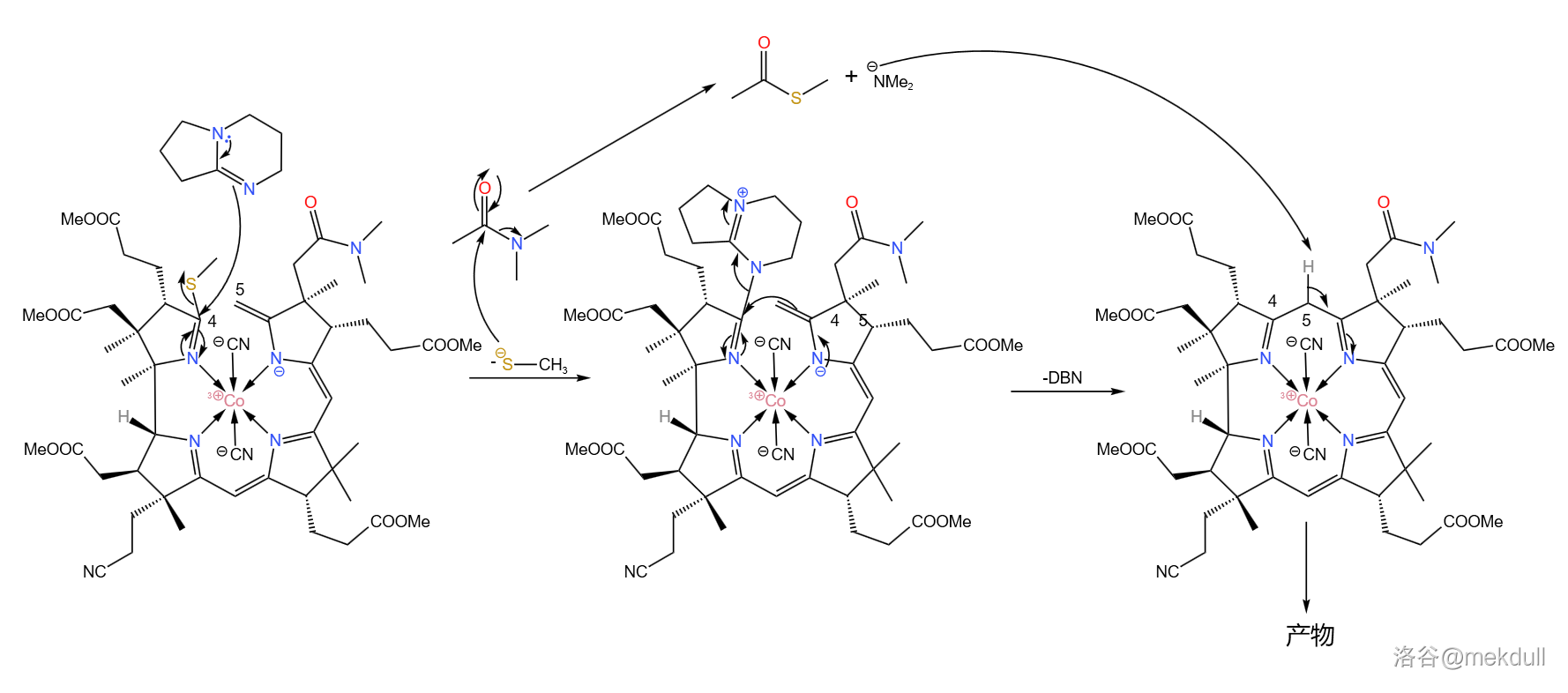
现在,整个咕啉环的拼接已经宣告成功,接下来便是对其进行修饰。在 $\ce{AcOH}$ 中用碘单质氧化该物质,$8$ 号位的 $\ce{C-H}$ 键断裂,分子右上角的 $7b$ 号位的酰胺结构变成内酯:
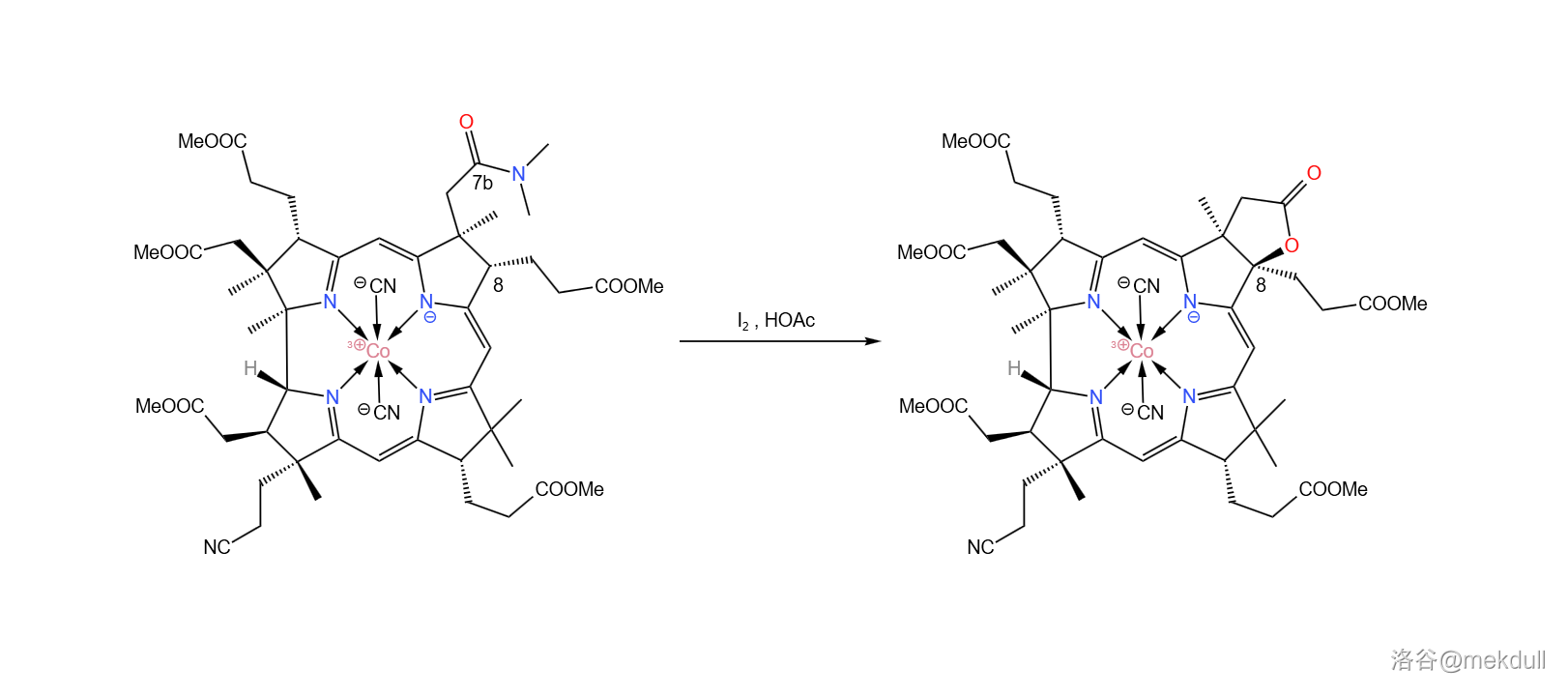
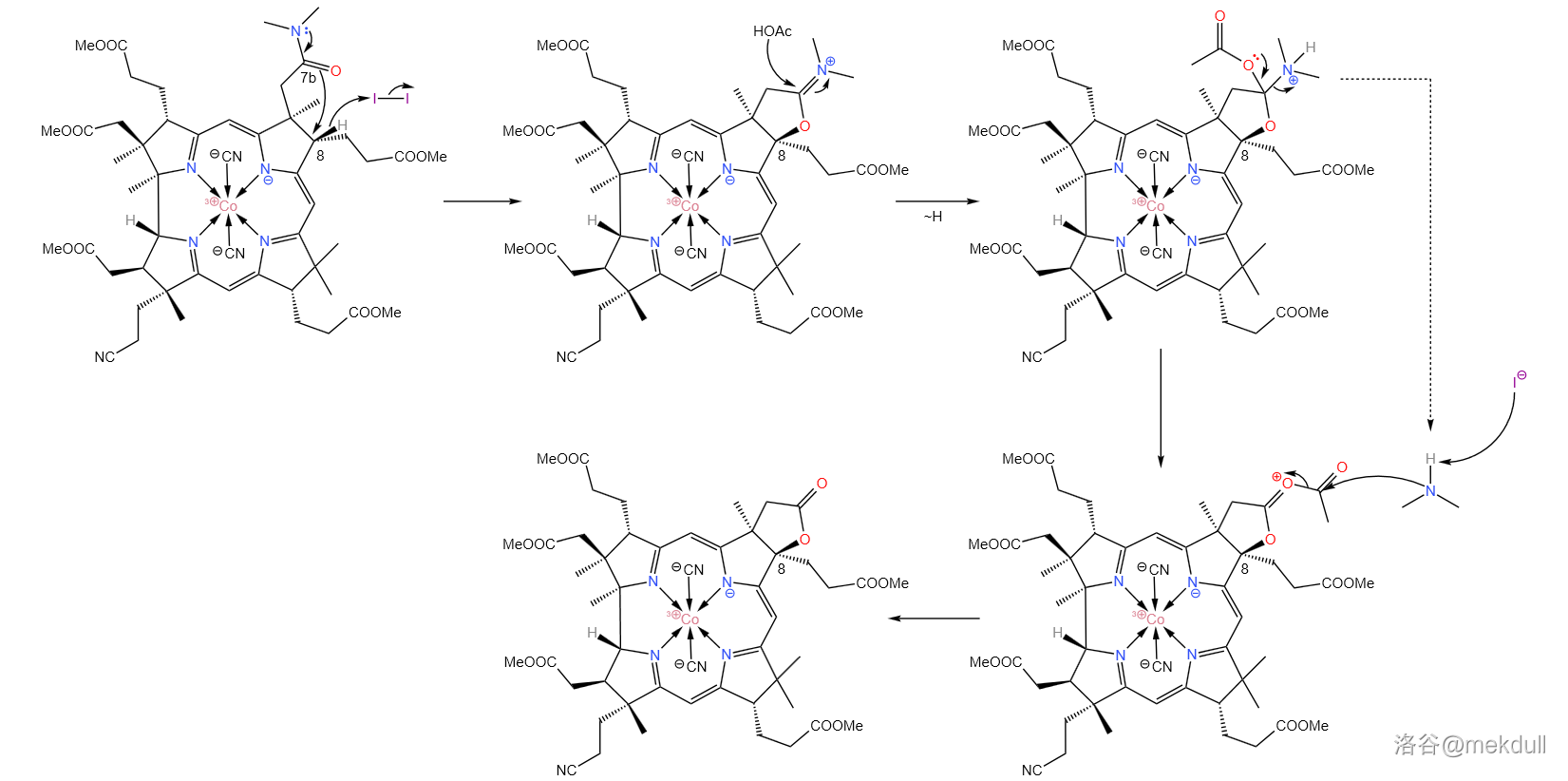
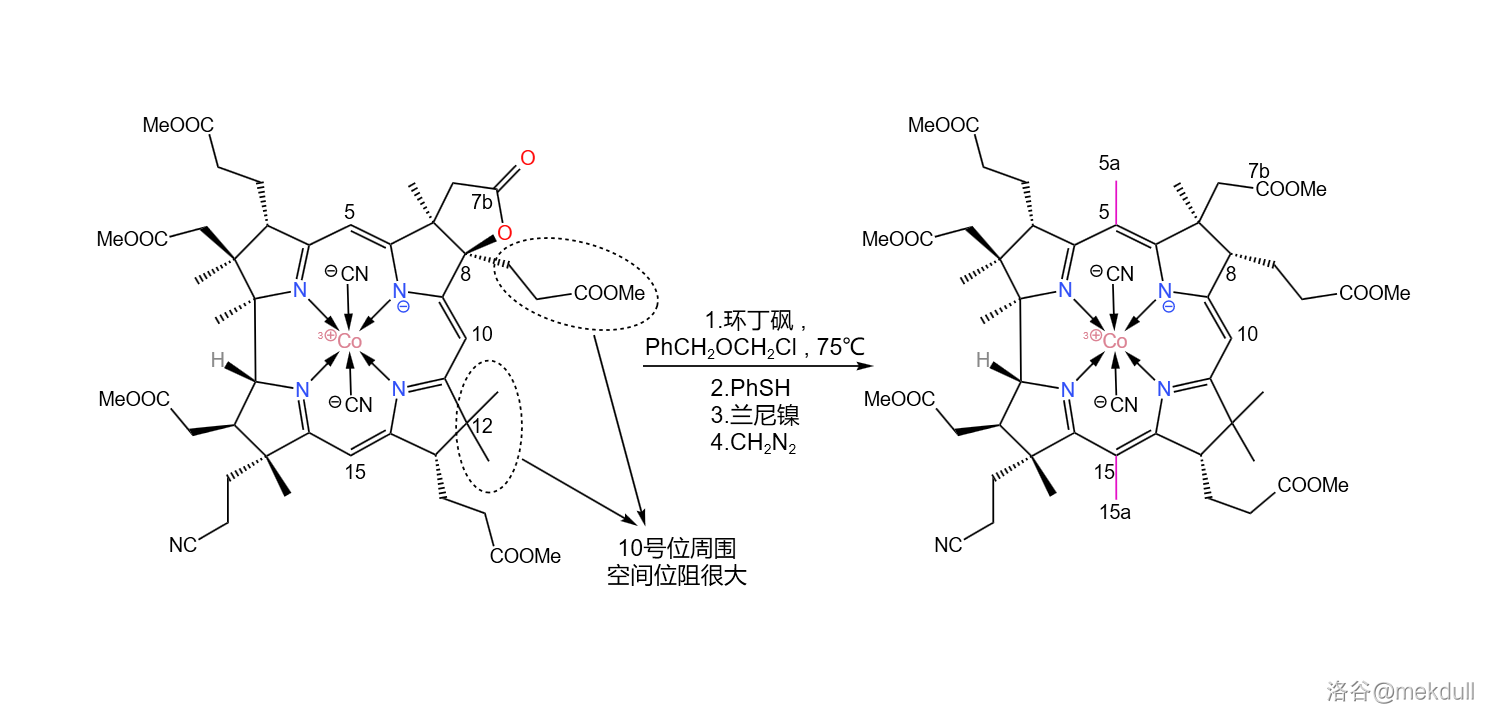
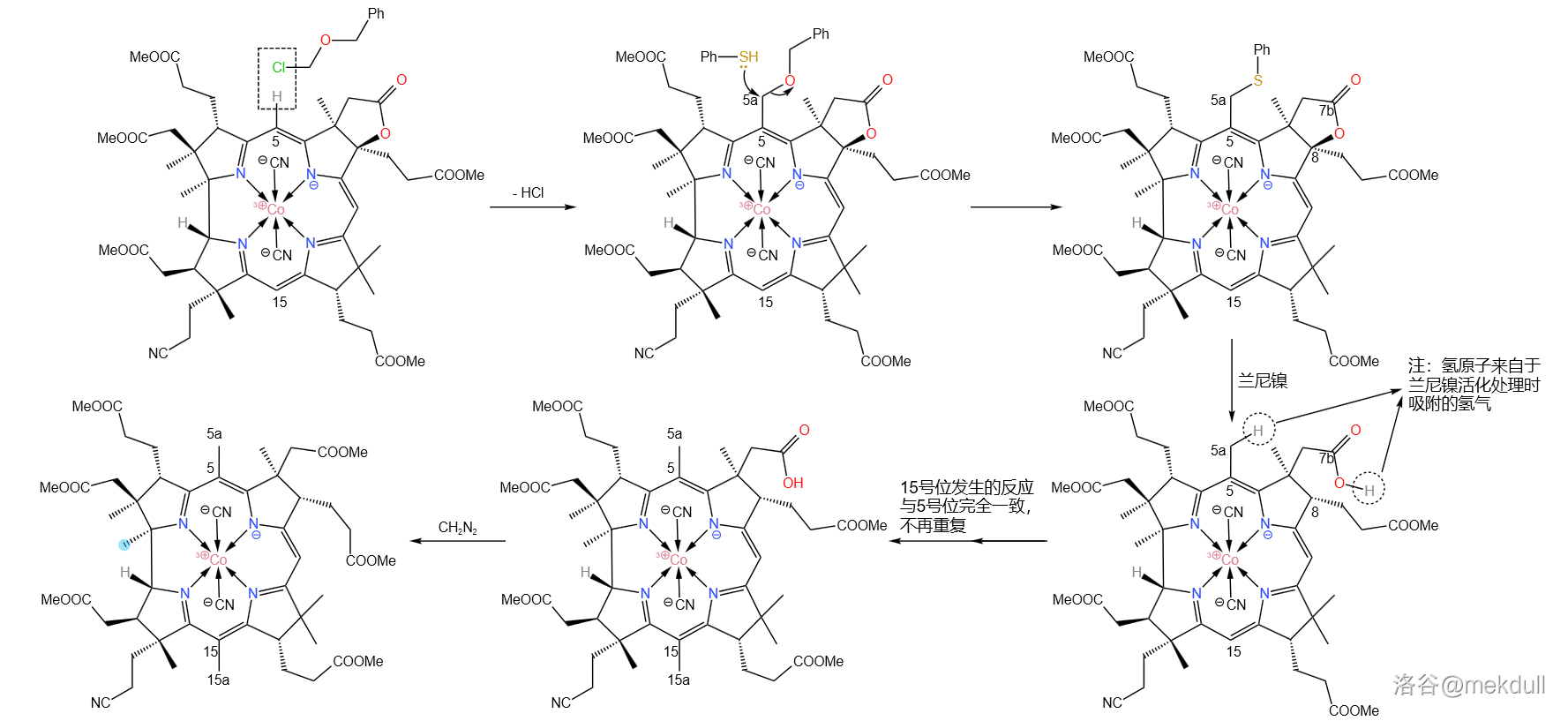
下一步,我们需要在 $5$ 号位和 $15$ 号位上各补上一个甲基(或许很多人都没有注意到这两个位置缺少一个甲基)。在环丁砜(溶剂)中,用苄基氯甲基醚($\ce{PhCH2OCH2Cl}$)可以直接活化 $5,15$ 号位上的 $\ce{C-H}$ 键并取代之;随后加入苯硫醇($\ce{PhSH}$),$5a,15a$ 号碳原子上的苄氧基($\ce{-OCH2Ph}$)随即被取代;接着,用兰尼镍($Raney~\ce{Ni}$)进行还原脱硫反应,$7b$ 号位上的内酯结构也随即被破坏,游离出羧基;最后用 $\ce{CH2N2}$ 进行酯化,完成整个反应历程。
有人可能会问,为什么 $10$ 号位上的氢原子没有被取代呢?观察分子的结构就能发现,$10$ 号位的周围的空间非常拥挤,附近的 $8,12$ 号位的碳原子都是完全取代的(其实,上一个反应的目的就是将 $8$ 号位完全取代,从而增大空间位阻),就像是一道屏障一般阻隔了试剂对 $10$ 号位的进攻。这足以保证这个反应的选择性:
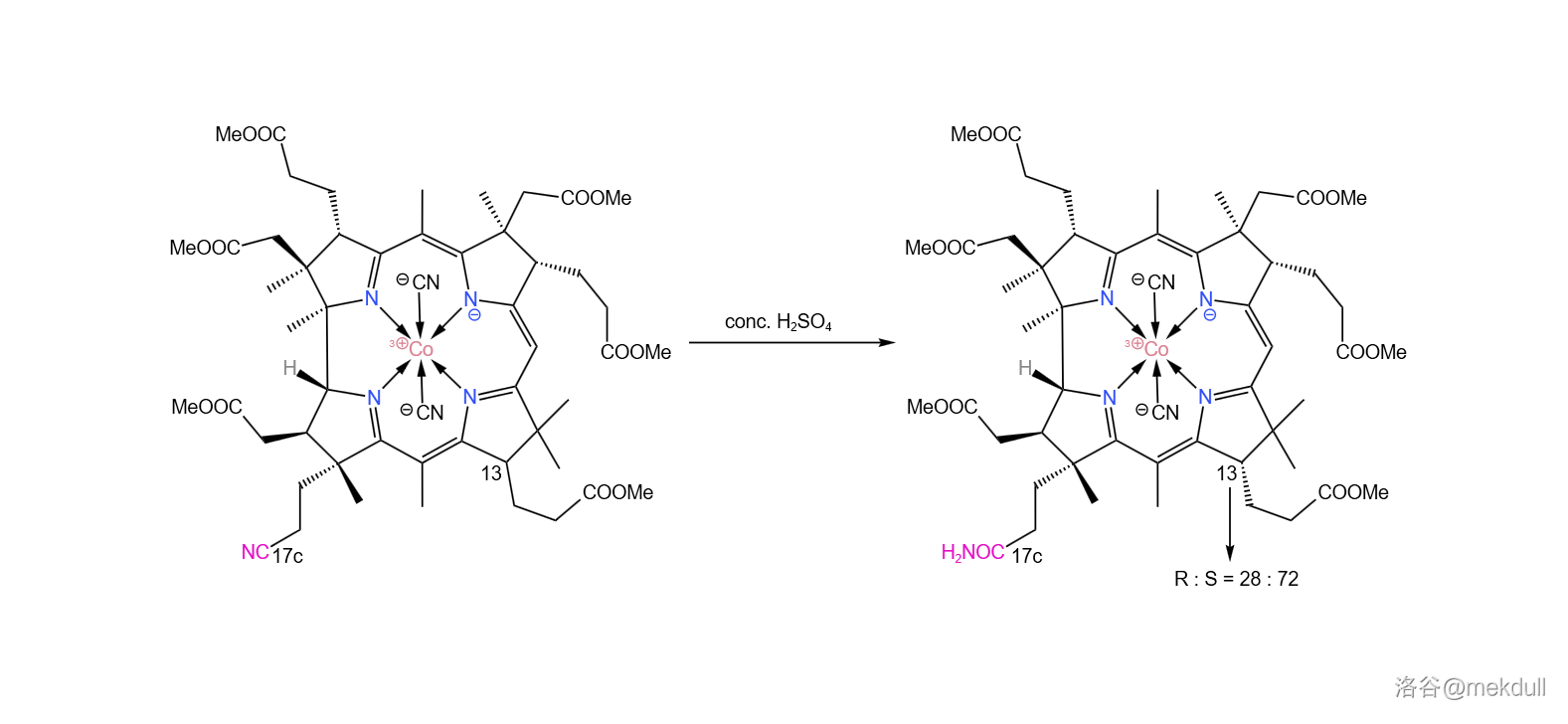
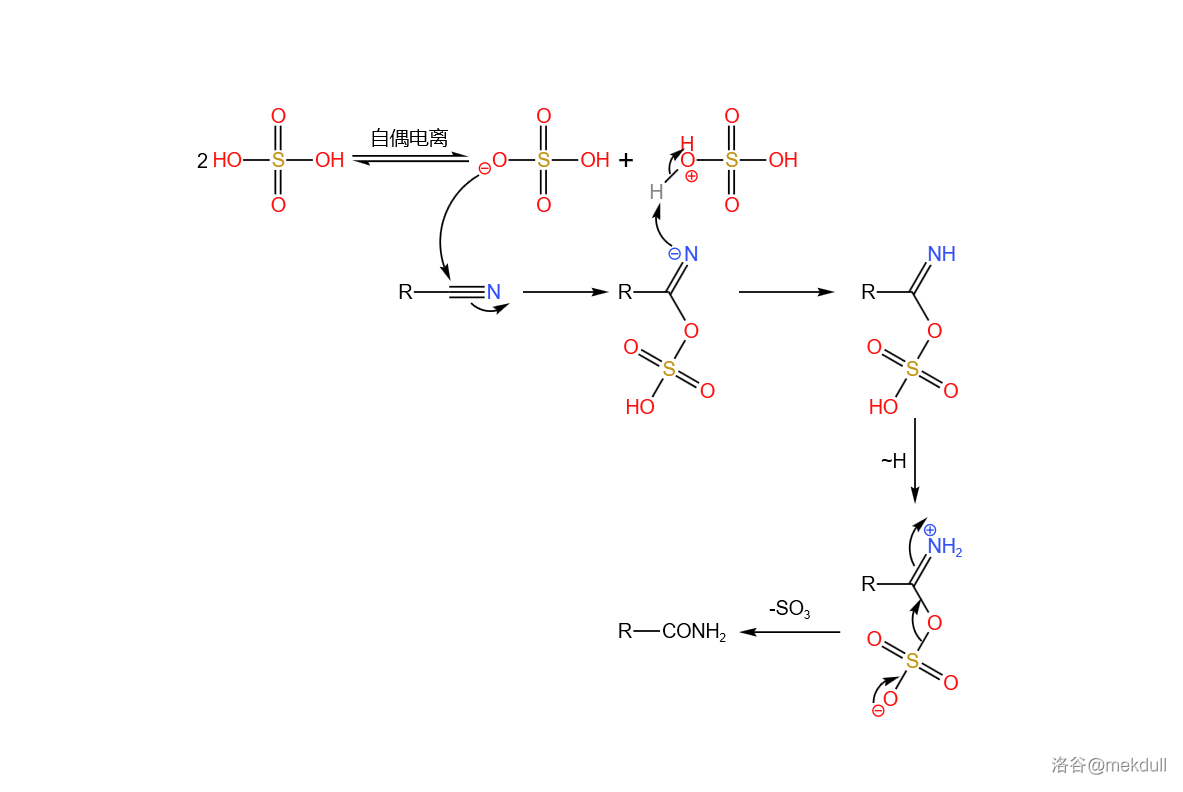
随后,我们需要把 $17c$ 号位的氰基($\ce{-C#N}$)水解成羧基($\ce{-COOH}$)。由于分子中有众多易水解的酯基,所以这并不是一项简单的任务。课题组选择先用浓硫酸处理该物质,将氰基水解为末端酰胺。没错,虽然一般浓硫酸是以“脱水剂”的身份出现的,但这里它扮演了一个相反的角色。这里的反应条件看似强烈,但主体的咕啉结构基本没有遭到破坏。
除此之外,浓硫酸还起到了一个意料之外的作用。分子中的 $13$ 号碳原子可以通过互变异构的方式转变自己的构型,而浓硫酸恰好提供了这个条件。最终,$13$ 号碳原子的 $R:S$ 平衡为 $28:72$(我们需要的是 $S$ 型):
接下来就是考虑如何将酰胺结构水解成羧基。为了防止酯基被水解,课题组首先考虑了使用亚硝酸酯(通式 $\ce{RONO}$)作为水解试剂。遗憾的是,亚硝酸酯虽然出色地完成了水解任务,但它同时也会将分子的 $10$ 号位亚硝化。为此,$Woodward$ 课题组筛选了众多亚硝酸衍生物,但都无济于事。最终,这一方案被放弃了。
在此关键时刻,隔壁的 $Eschenmoser$ 课题组送来了助攻。简单来说,他们发明了一种强大的亲电试剂(在此就姑且称之为 $Eschenmoser$ 亲电试剂吧),可以通过氟硼酸银($\ce{AgBF4}$)与一种环己胺衍生物反应生成。在非常弱的酸性条件下(可以用酸碱缓冲体系),它就可以将酰胺转化为羟乙醛酯。最后只需加入 $\ce{Me2NH}$,就可以将活泼的羟乙醛酯转化为羧酸:
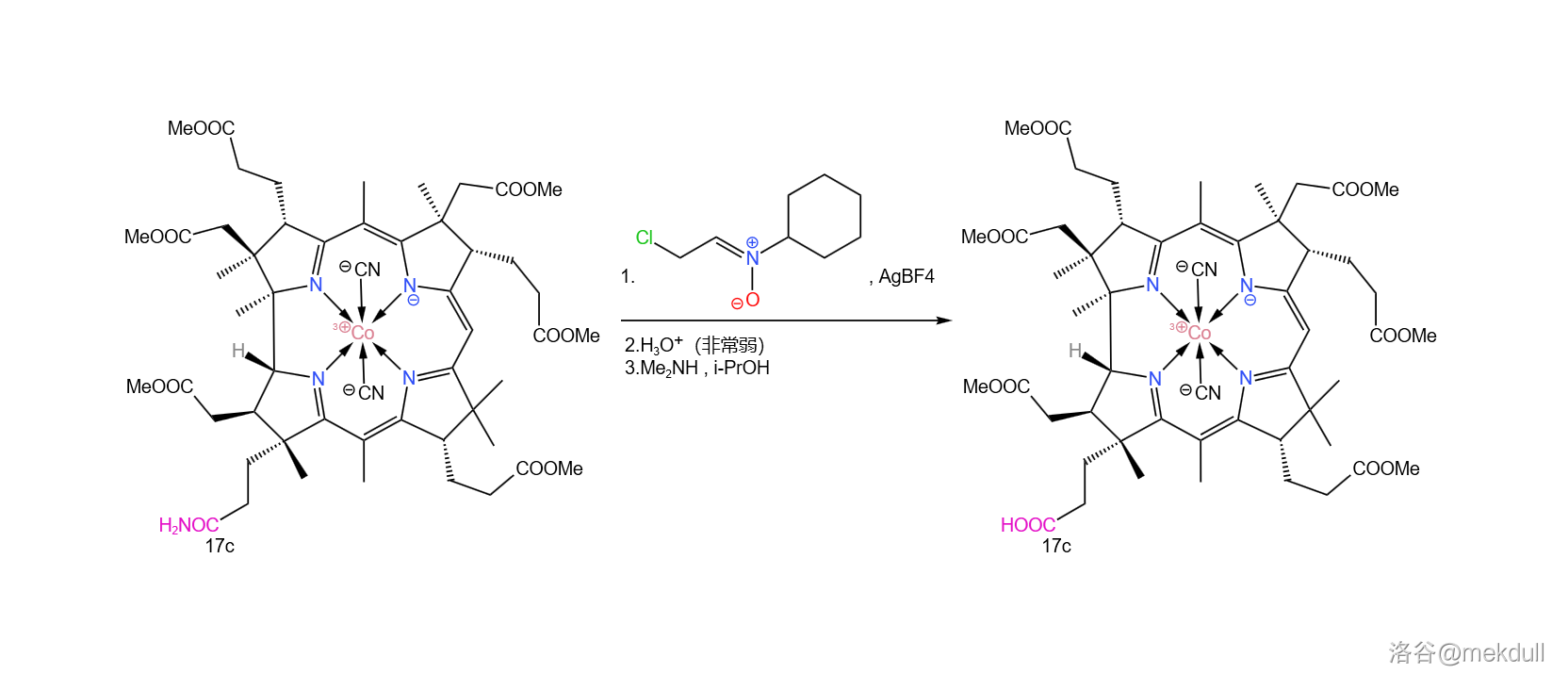
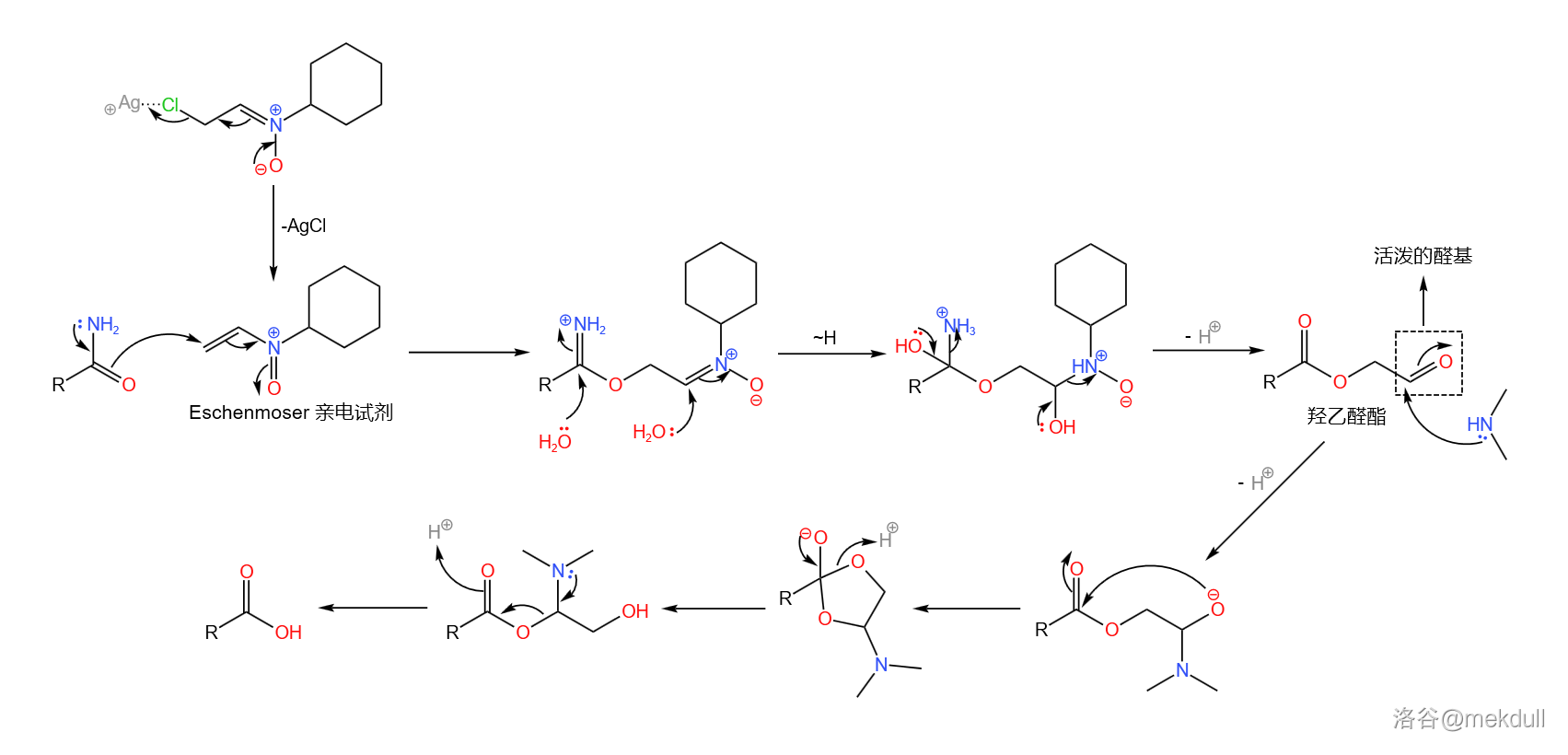
这个反应的设计极其巧妙,而且在面对如此复杂的底物时也可以给出超过 $90\%$ 的产率——$Woodward$ 评价这个反应“狡猾而优雅”。后续的研究证明,这个反应对于几乎所有酰胺都是有效的,不仅限于末端酰胺。如今,我们一般把它称为 $Eschenmoser$ 酰胺水解反应,在复杂天然产物的合成中常会用到。
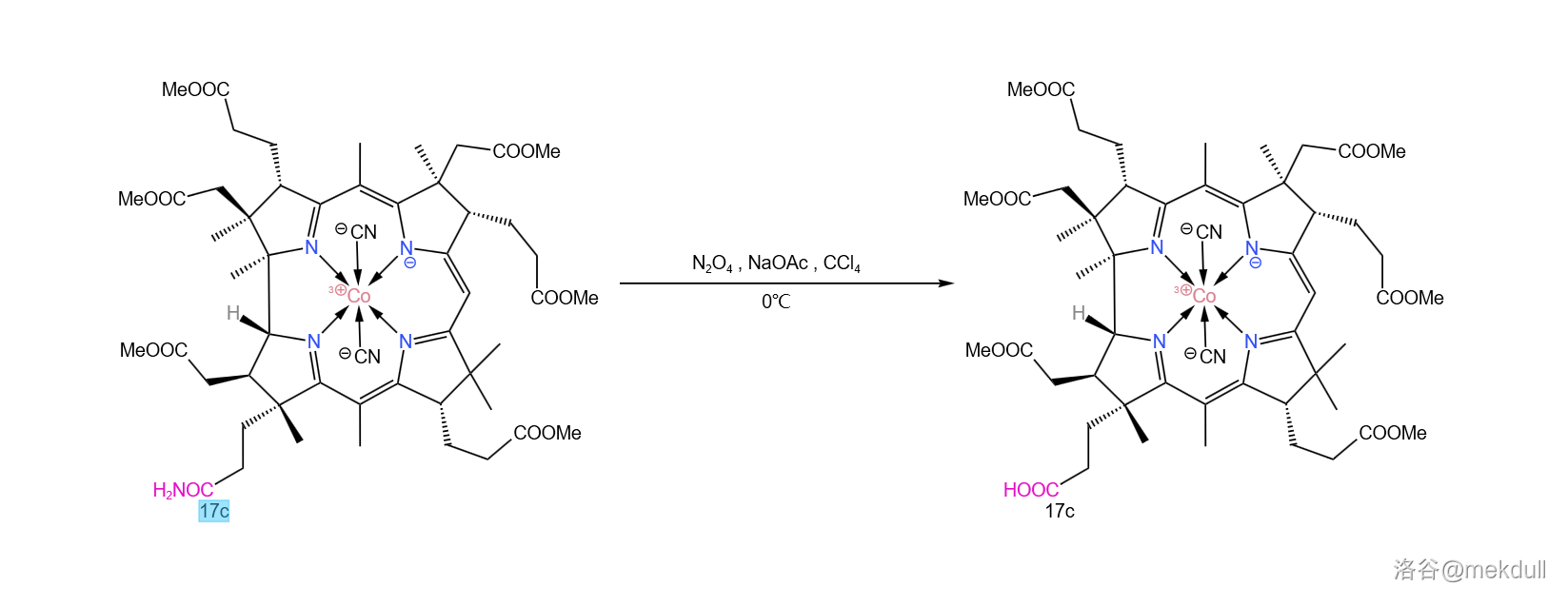
或许是不愿落后于人,$Woodward$ 课题组很快也发现了这个问题的另一种解法:在四氯化碳($\ce{CCl4}$)中用 $\ce{NaOAc}$ 和四氧化二氮($\ce{N2O4}$)代替亚硝酸酯,就可以将酰胺有效水解,同时分子的 $10$ 号位不再被亚硝化。反应产率 $\ce{70\%-80\%}$,虽然比 $Eschenmoser$ 的方法差了点,但如果从经济上考虑,那 $Woodward$ 的方法显然是更优的:
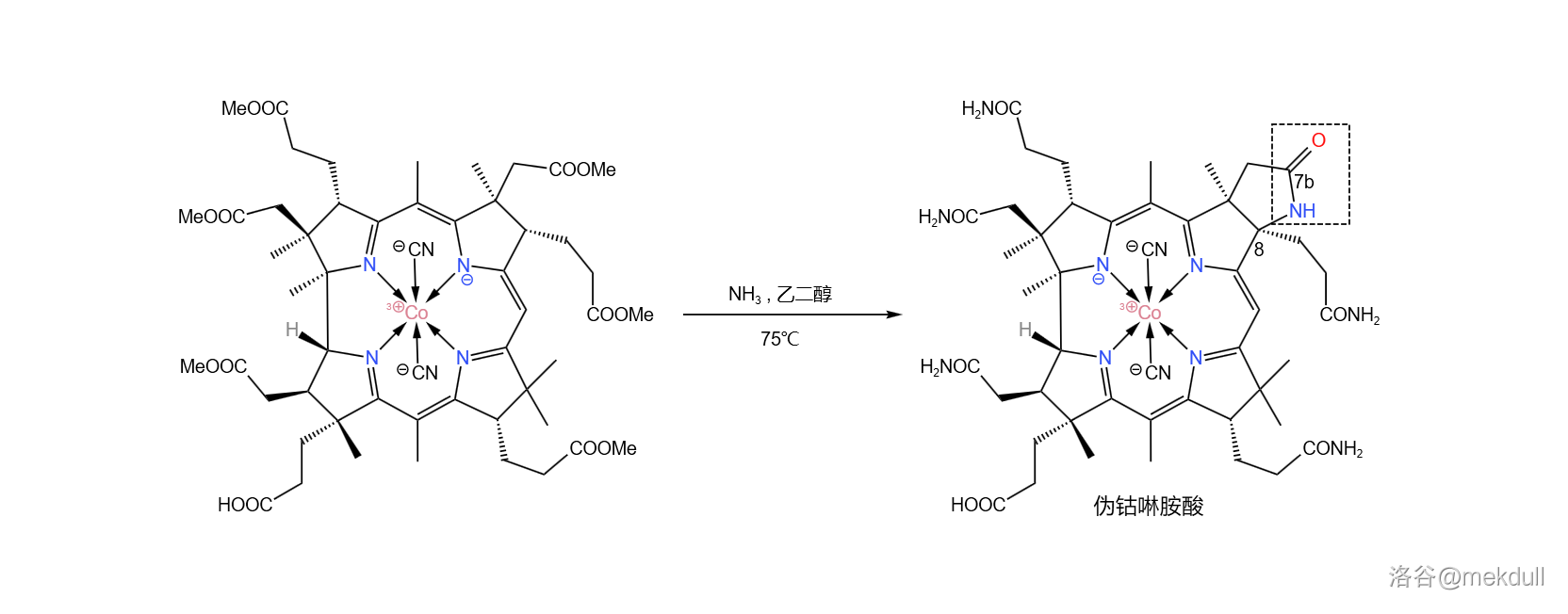
现在,距离成功合成钴啉胺酸只差一步了:将分子中的六个酯基全部转化为酰胺。在 $75^{\circ}C$ 左右用氨($\ce{NH3}$)与乙二醇($\ce{HOCH2CH2OH}$)大致 $1:1$ 的混合物处理该物质,可以以高产率得到一种结晶。但令人大跌眼镜的是,它根本不是钴啉胺酸!
这种物质的化学性质与钴啉胺酸几乎一致,且两者具有基本相同的红外光谱、电子光谱,甚至于连纸色谱、平面色谱都完全无法区分二者。只有高压液相色谱系统告诉我们:它们不一样!如下图所示,课题组得到的物质在化学式上比钴啉胺酸少了两个 $\ce{H}$。在它的右上角,$7b$ 号碳原子形成了内酰胺结构。课题组将这种物质称为“伪钴啉胺酸”($pseudocobyric~acid$):
为什么分子会莫名其妙地去掉两个 $\ce{H}$?难道是有空气混进了反应体系?在反复检查之后,课题组排除了这种可能。那它到底为什么会被氧化呢?等等,分子中心还有一个配位的 $\ce{Co}$ 原子!$Woodward$ 猜想,中心配位的 $\ce{Co(III)}$ 可以通过某种机制变成 $\ce{Co(I)}$。由于 $\ce{Co(I)}$ 具有很强的还原性,所以它会迫使分子会放出 $\ce{H2}$,并把自己变回 $\ce{Co(III)}$。
所以,该怎么解决这个问题?一开始,课题组希望找到一种合适的还原剂,将伪钴啉胺酸还原成钴啉胺酸。他们搜寻了好几个月,但根本没有收获。就在此时,他们意外地发现,只需将少量铵盐(比如氯化铵 $\ce{NH4Cl}$)加入反应体系,效果就立竿见影。伪钴啉胺酸从产物中消失了,取而代之的是几乎 $100\%$ 产率的钴啉胺酸:
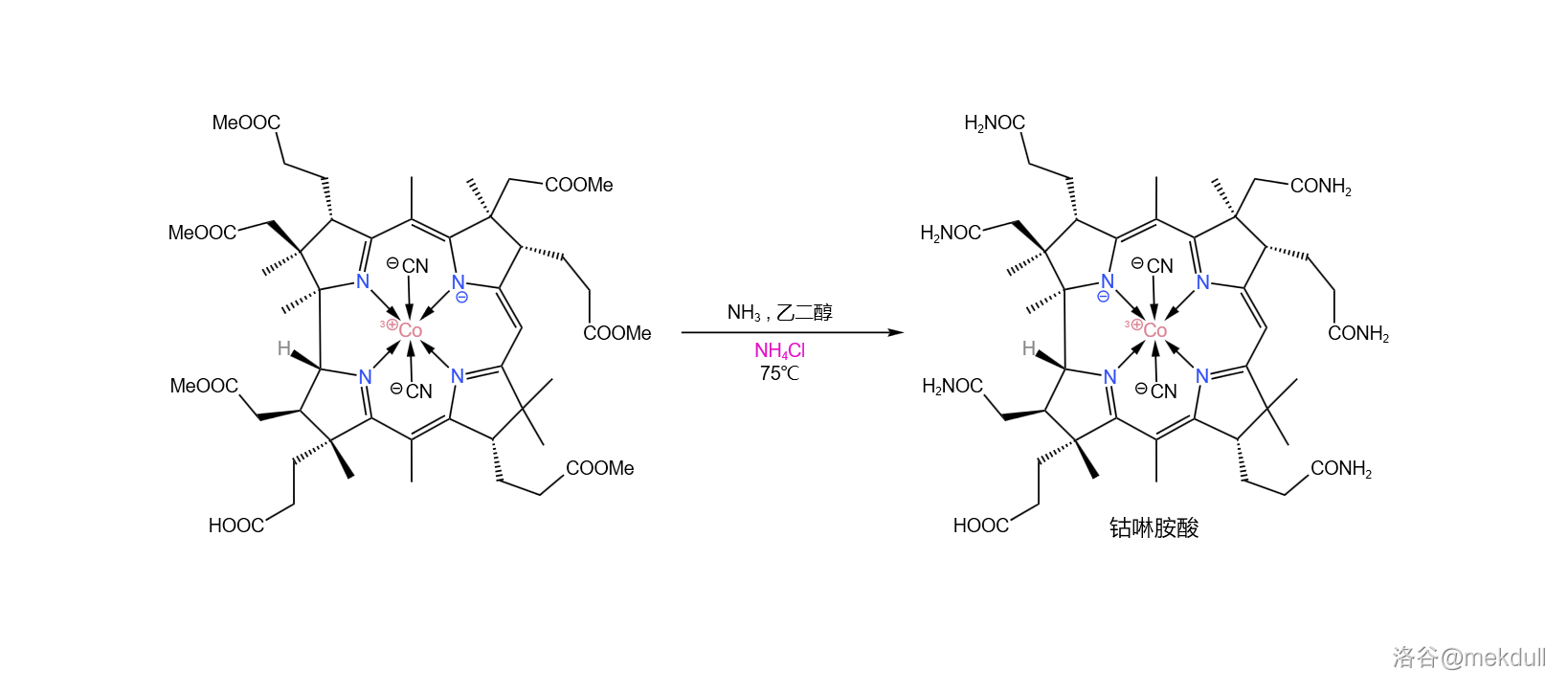
为什么铵盐会有这么神奇的效果?$Woodward$ 认为,生成伪钴啉胺酸的机理中可能包含一个酰胺负离子中间体($\ce{-CONH^-}$),而铵根离子($\ce{NH4+}$)可以有效地淬灭酰胺负离子,从而抑制了伪钴啉胺酸的生成。
由于没有详细研究,$Woodward$ 并没有在论文中给出生成伪钴啉胺酸的详细机理。但这或许已经不重要了,因为这一步的完成,标志着维生素B12的全合成宣告成功。这座“有机合成界的珠穆朗玛峰”,就这样在人们的齐心协力之下被征服了。这一年是 $1972$ 年。
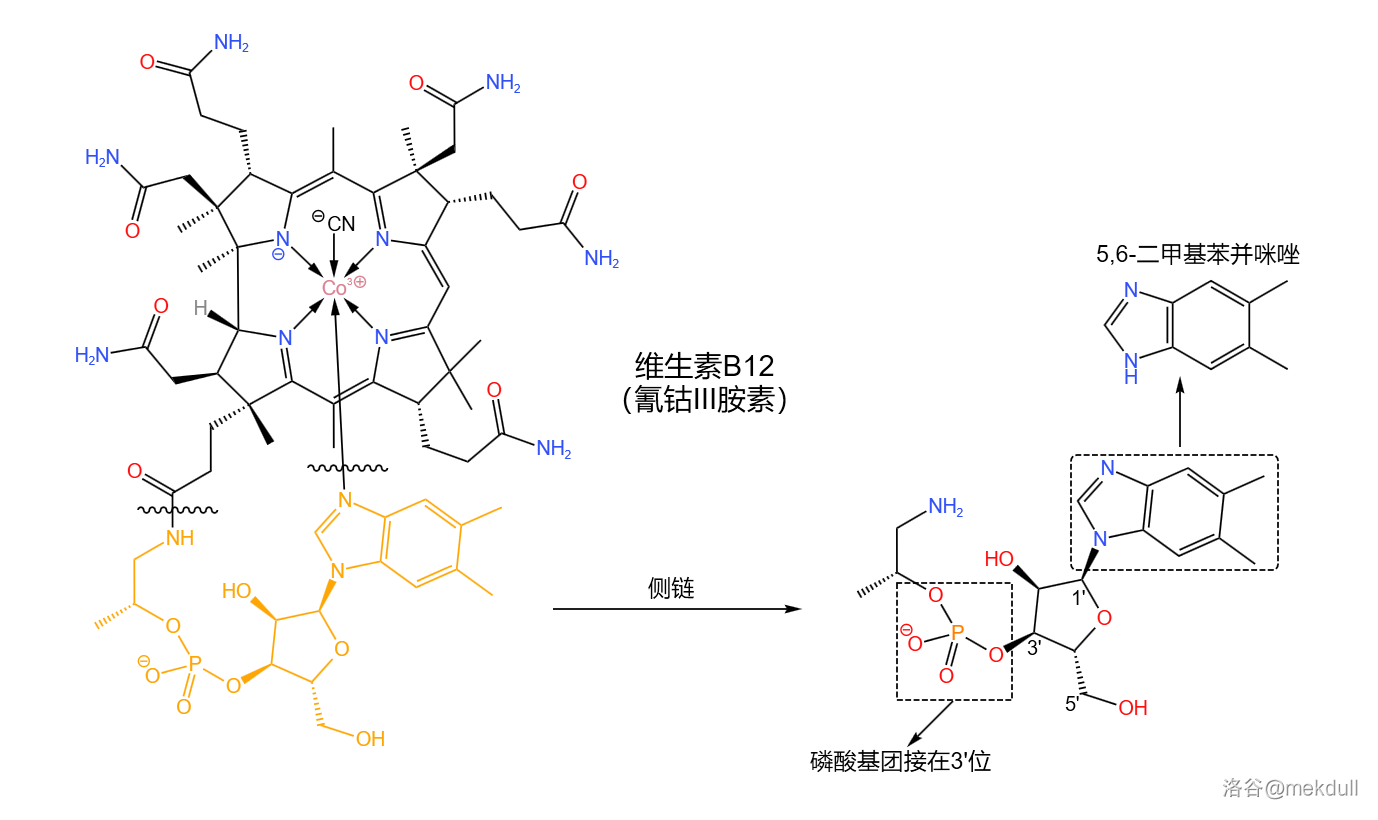
但是,我们的旅程还没有结束。毕竟,钴啉胺酸距离维生素B12还差一个结构类似于核苷酸的侧链,那这个侧链又应该如何合成呢?
3.4 核苷酸侧链的合成与拼接
在概述中已经提到,这一部分的工作是德国化学家 $Bernhauer~K.$ 等人在 $1960$ 年完成的,所以对于 $Woodward$ 来说,这部分的合成方法是现成的。但无论怎么说,这条侧链都是维生素B12的一部分,所以一篇完整的全合成介绍是少不了它的。
由于侧链的结构类似于核苷酸,所以 $Bernhauer$ 的课题组也采用了类似于核苷酸的合成策略。但值得注意的是,这条侧链的“碱基”并不是 $\ce{A,C,G,T,U}$ 中的任何一个,而是一种苯并咪唑;磷酸基团则是接在核糖结构的 $3’$ 位,而不是 $5’$ 位:
合成的起始物是一种有机染料工业的重要原料:$4,5-$二甲基邻苯二胺。在多聚磷酸(或其他较强的酸)的作用下,它可以与甲酸缩合得到 $5,6-$二甲基苯并咪唑:
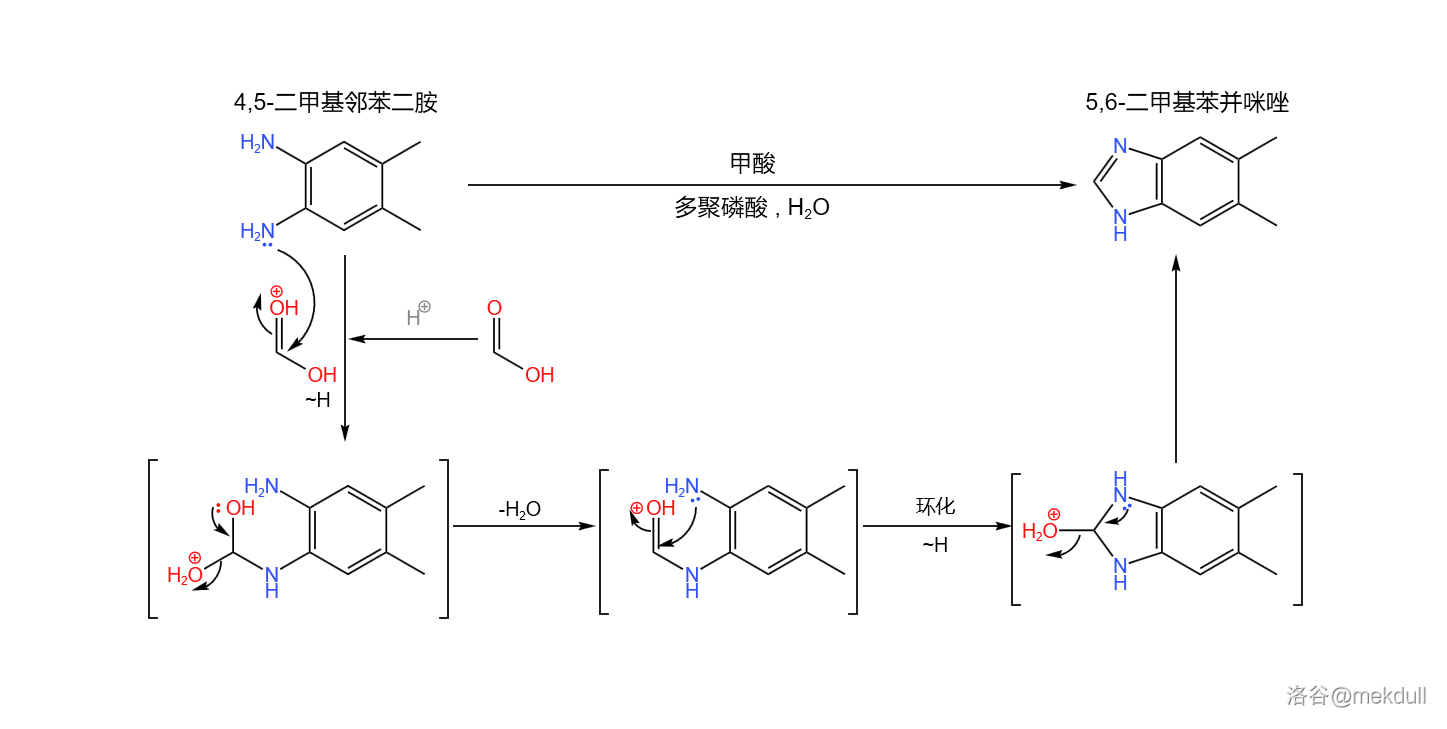
(注:较新的研究发现,上反应可以在 $\ce{DMF}$ 中被三甲基氯硅烷($\ce{TMSCl}$)催化,在此条件下产率可以达到 $95\%$。上图中展示的反应条件是上世纪使用的,产率相对较低)
这样一来,碱基就搞定了。随后登场的是一种核糖的衍生物:四乙酰核糖——它直到今天都是核苷酸合成中的重要试剂。在二甲苯中,它与刚才制得的 $5,6-$二甲基苯并咪唑在 $1’$ 号位发生缩合反应;将反应的产物用 $\ce{NH3-CH3OH}$ 处理,脱去多余的乙酰基后得到结构类似于核苷的物质:

下一步,在低温下用三氯氧磷($\ce{POCl3}$)和磷酸三乙酯($\ce{PO(OEt)3}$,做溶剂)处理该物质,得到 $5’$ 号位的二氯磷酸酯结构;随后用强碱处理之,得到 $3’,5’$ 号位的环状磷酸盐:
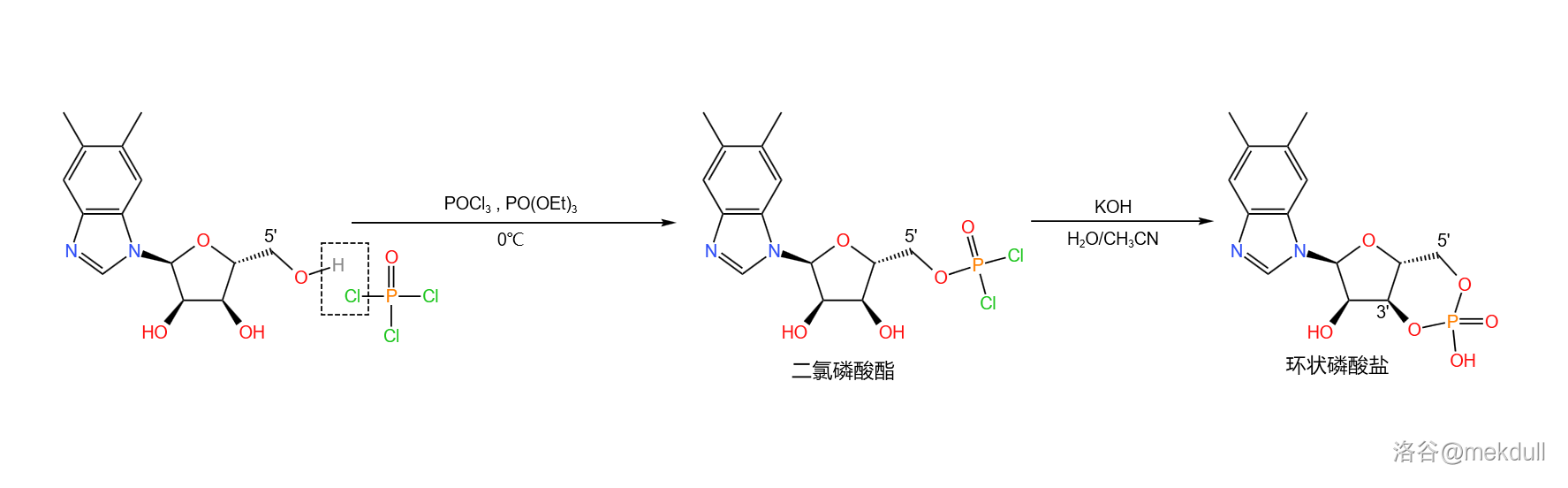
接着,在很弱的碱性($\text{PH}\approx8.0$)条件下,用铈离子($\ce{Ce^3+}$,此处由于碱性太弱无法产生氢氧化铈($\ce{Ce(OH)3}$)沉淀;最近有研究指出,实际上起作用的是 $\ce{Ce^3+}$ 在此条件下部分水解产生的簇状羟基配合物)催化环状磷酸盐部分水解,可以得到 $3’$ 号位的磷酸(副产 $5’$ 位的磷酸):
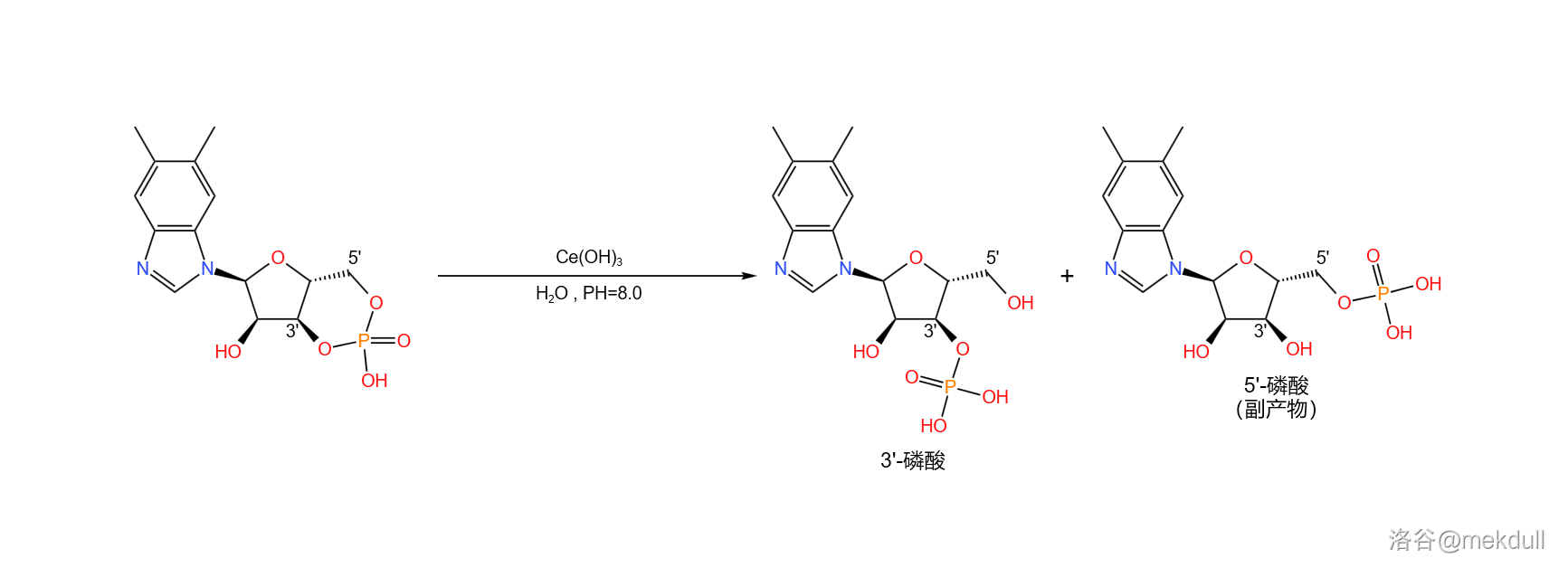
下一步,我们要把 $3’-$ 磷酸与异丙醇胺($\ce{H2NCH2CHOHCH3}$,有两种旋光异构体,此处使用 $R-$异丙醇胺)缩合成磷酸酯。为了防止氨基的干扰,课题组首先将其与苯甲醛($\ce{PhCHO}$)反应,把氨基保护起来;随后,在强碱正丁醇钠($\ce{t-BuONa}$)的作用下,两者顺利地发生缩合反应;反应结束后用乙酸水溶液脱去保护基,就能得到这条完整的核苷酸侧链:
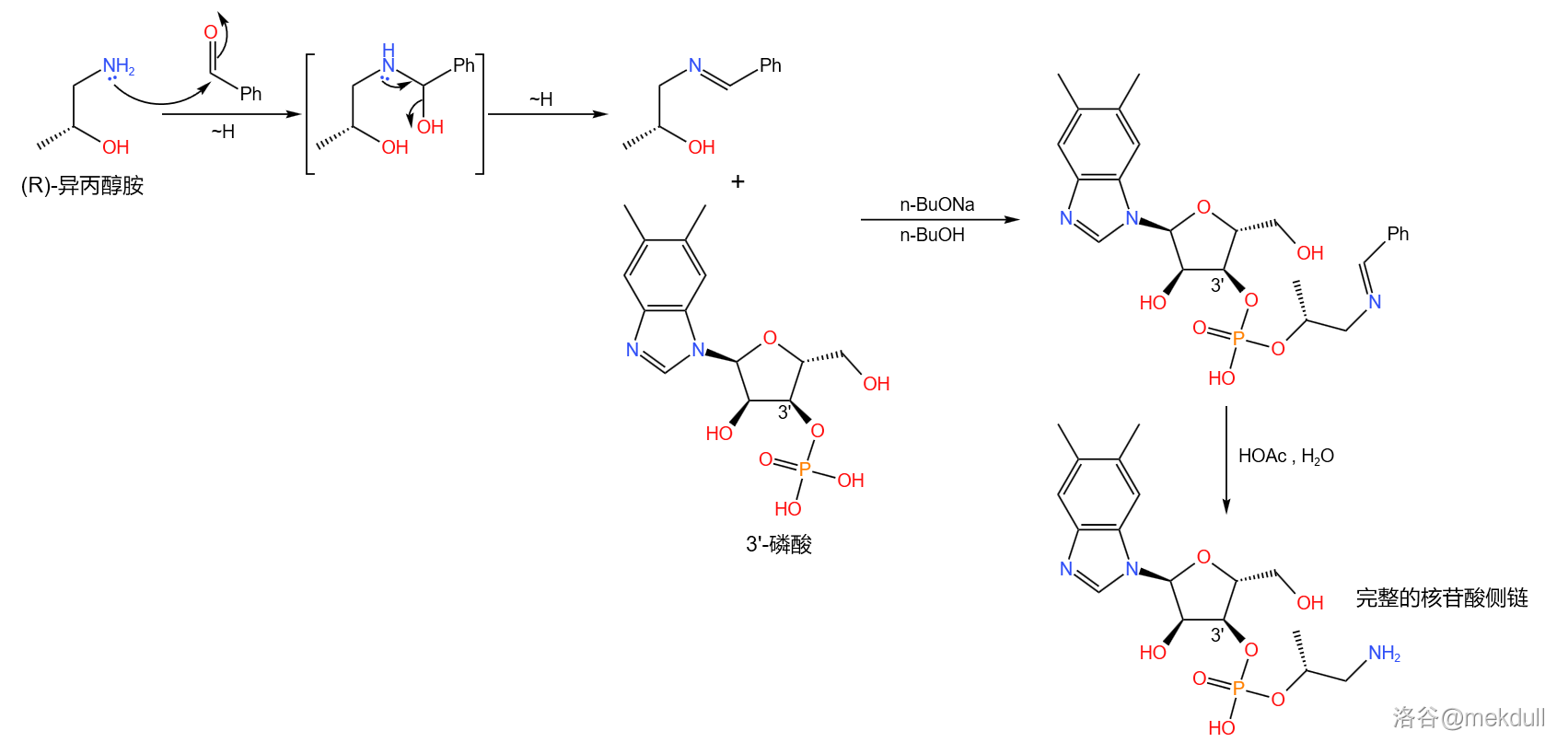
最后,我们需要把这条侧链“装”到钴啉胺酸上。为此,课题组测试了多种反应条件,最终选择使用柠檬酸铵溶液和异戊醇的混合物处理。二者顺利地发生脱水缩合反应,以 $92\%$ 的产率得到最终产物:
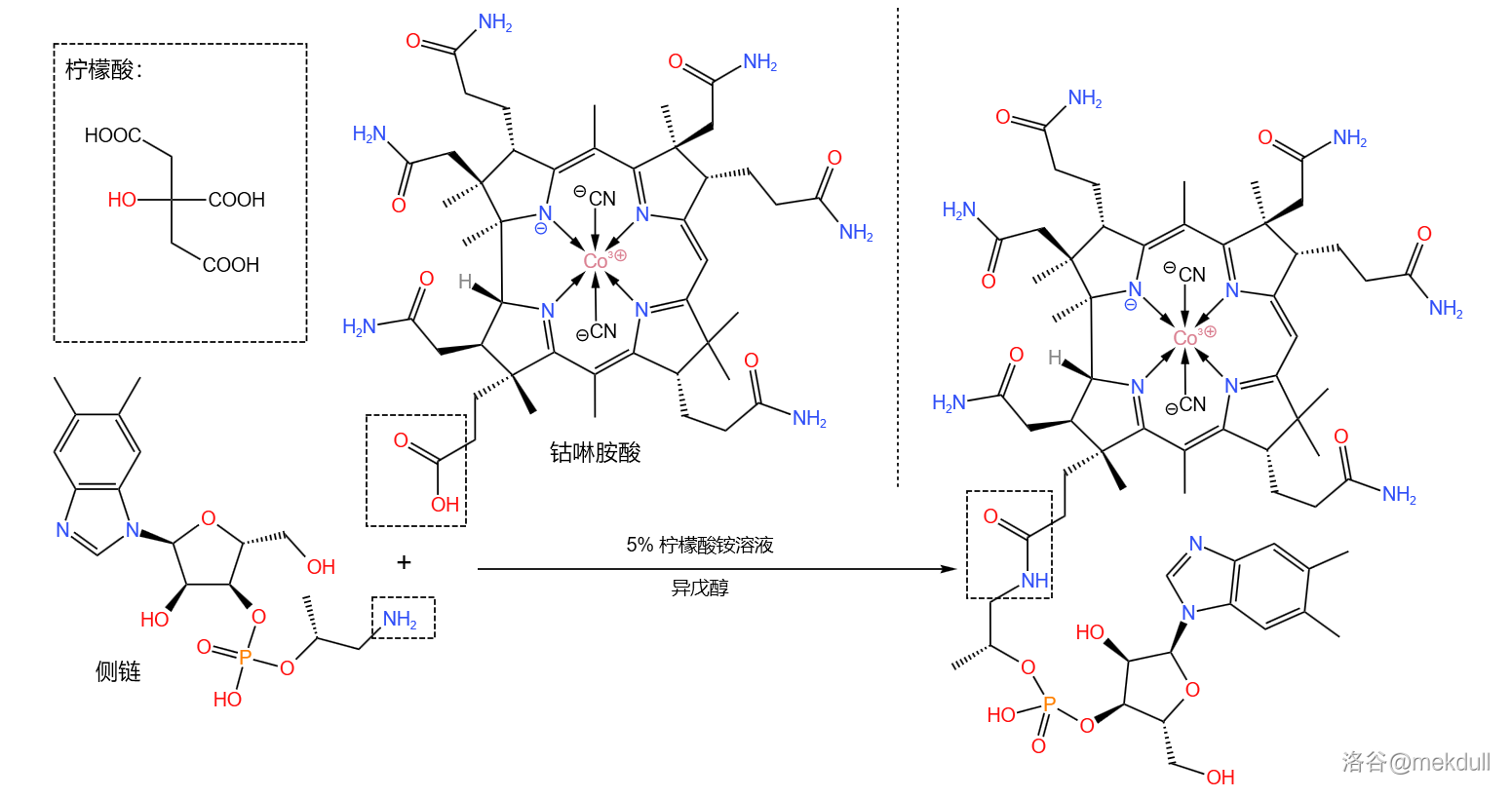
随后,该物质在此条件下可以自发消去一个 $\ce{HCN}$ 分子,而侧链顶端的 $\ce{N}$ 原子则会自动进入中心 $\ce{Co}$ 原子的配位体系,从而得到成品维生素B12:
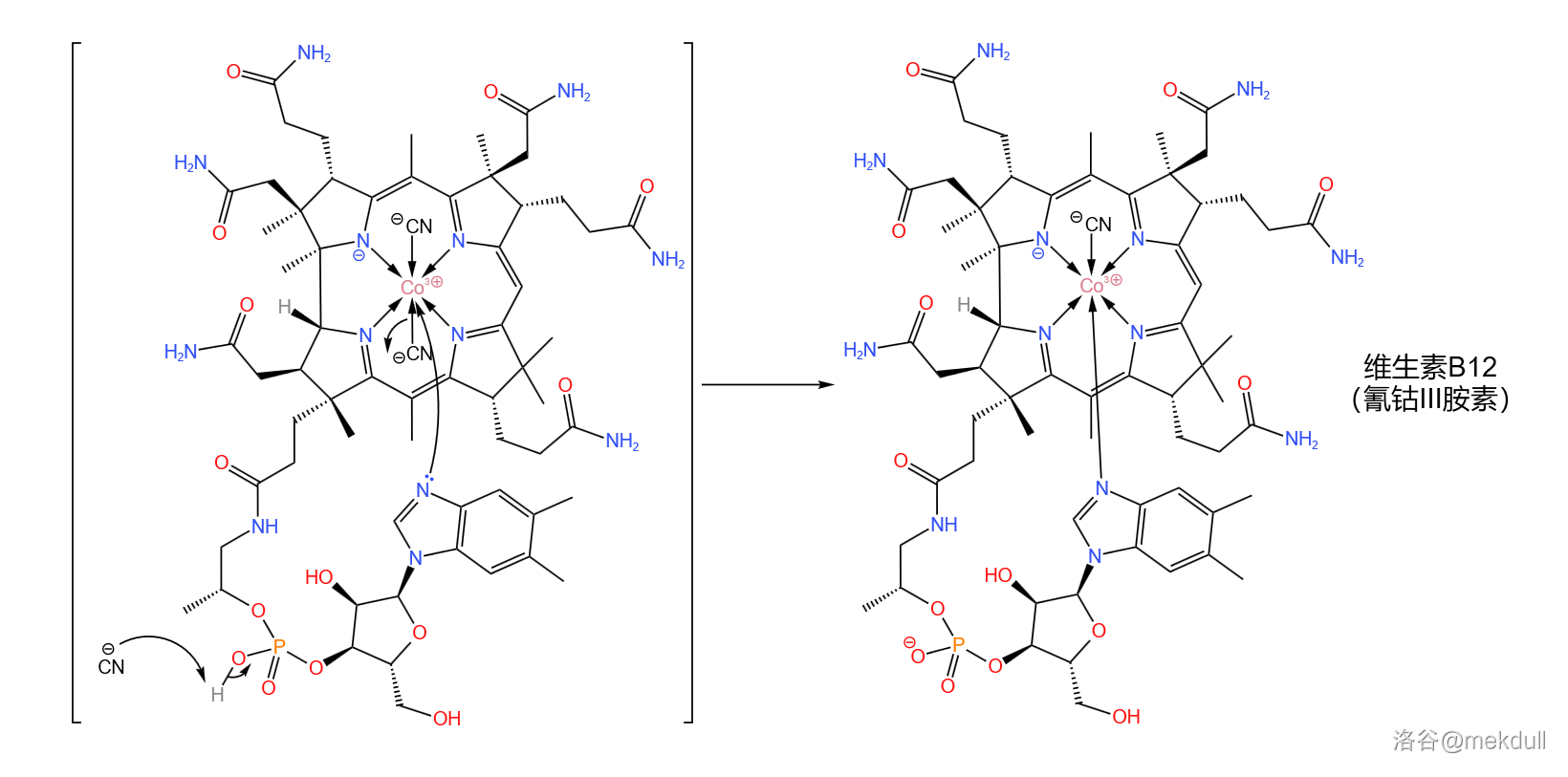
至此,那一抹亮丽的红色终于出现在了人们眼前。
4.总结与后记
维生素B12的全合成是有机合成界前所未有的壮举。毫不夸张的说,这项工作可以成为划分“近现代”有机化学和“当代”有机化学的分界线。它的意义远不止拓展了有机合成复杂性的界限,更重要的是,它的成功沉重打击了几百年来“化学局限论”的思想,让人们开始相信:化学真的可以为人类创造一个新的世界。
即使抛开思想上的启迪,这项工作在实际上的意义也不遑多让。在此次全合成中首次亮相的 $Eschenmoser$ 偶联反应已经成为构筑碳碳键的重要手段,在许多药品的工业化合成中得到应用;$Bernhauer~K.$ 等人合成侧链的方法在之后的岁月中被不断改进,衍生出了众多重要的核苷酸化学合成法,为今日腺苷、肌苷、$\ce{ADP}$、$\ce{ATP}$ 等生物分子的大规模合成打下了基础,也成为了遗传物质化学合成的支点之一;咕啉环的人工合成法指导了人们探寻生物体内咕啉结构的构建,并最终将其合理化,阐明了生物合成维生素B12的机理;$Woodward$ 团队在合成过程中阐明了互变异构导致的构型翻转,再一次提升了人类对于手性的认识,指导着后世几乎所有的手性合成方法;合成过程中发现的 $\ce{Co}$ 元素的显著催化作用,到今天已经发展成为有机合成的一种强大策略,诸如 $\ce{Co}$ 催化的偶联反应、环加成反应、消去反应等令人眼花缭乱;合成过程中,$Woodward$ 和他的学生 $Hoffmann$ 发现了 $\ce{D-A}$ 反应在不同条件下的立体选择性,两人进而将这些规律总结、归纳,最终提出了名震整个有机化学界的轨道对称守恒原理,也叫 $Woodward-Hoffmann$ 规则——这被认为是近年来化学理论方面的最大成就。
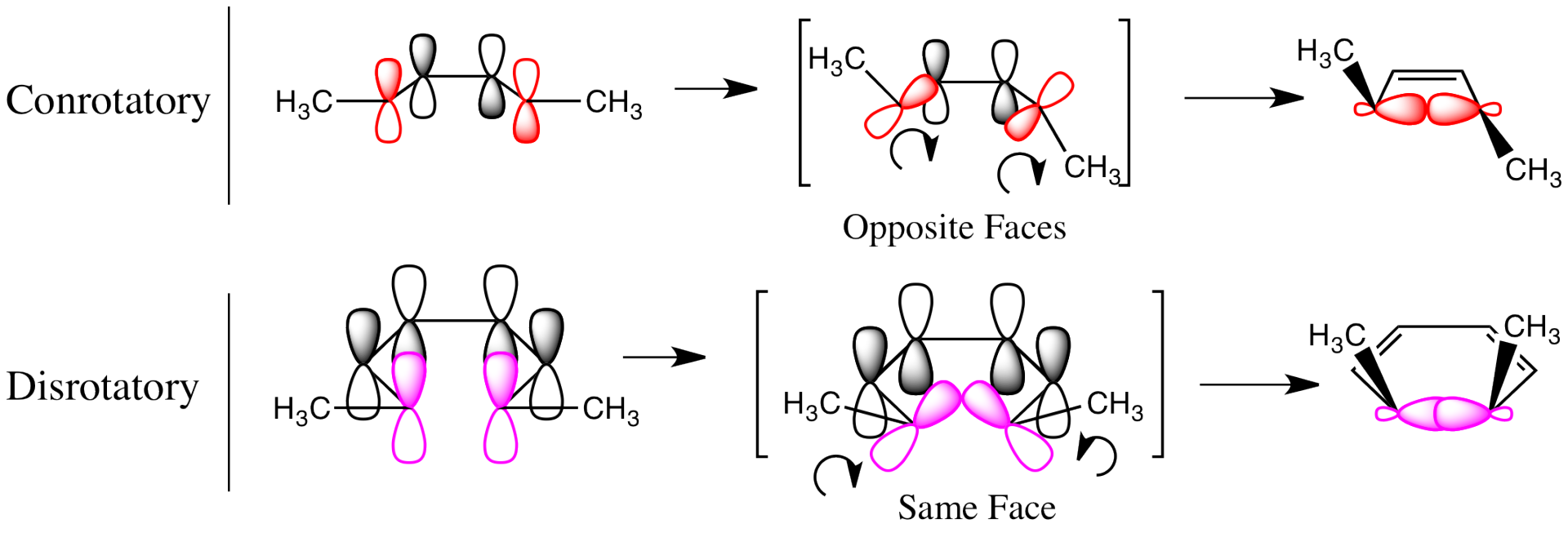
(上图:轨道对称守恒原理的简单图示)
在科学家们的努力之下,$1993$ 年,维生素B12的生物合成机制被基本阐明。在此之后,人们开发出了维生素B12的生物合成技术,这才有了今天能够直接在药店里买到的维生素B12药片。然而,谁能想到,在这种不起眼的廉价药品背后,暗藏着 $150$ 余年来无数科学家为之不懈奋斗的史诗。
$1979$ 年,积劳成疾的伍德沃德因心脏病突发去死,享年 $62$ 岁。他在去死的前一天仍念念不忘那还未完成的赤霉素($gibberellin$)全合成。最终,他的学生带领着剩下的课题组成员完成了这项工作;而在他去世 $15$ 年后,他的另一位学生 $Y.Kishi$ 将完成有机合成界“新的珠穆朗玛峰”——岩沙海葵毒素($palytoxin$)的全合成。如果伍德沃德能看到,想必他一定会倍感欣慰吧。
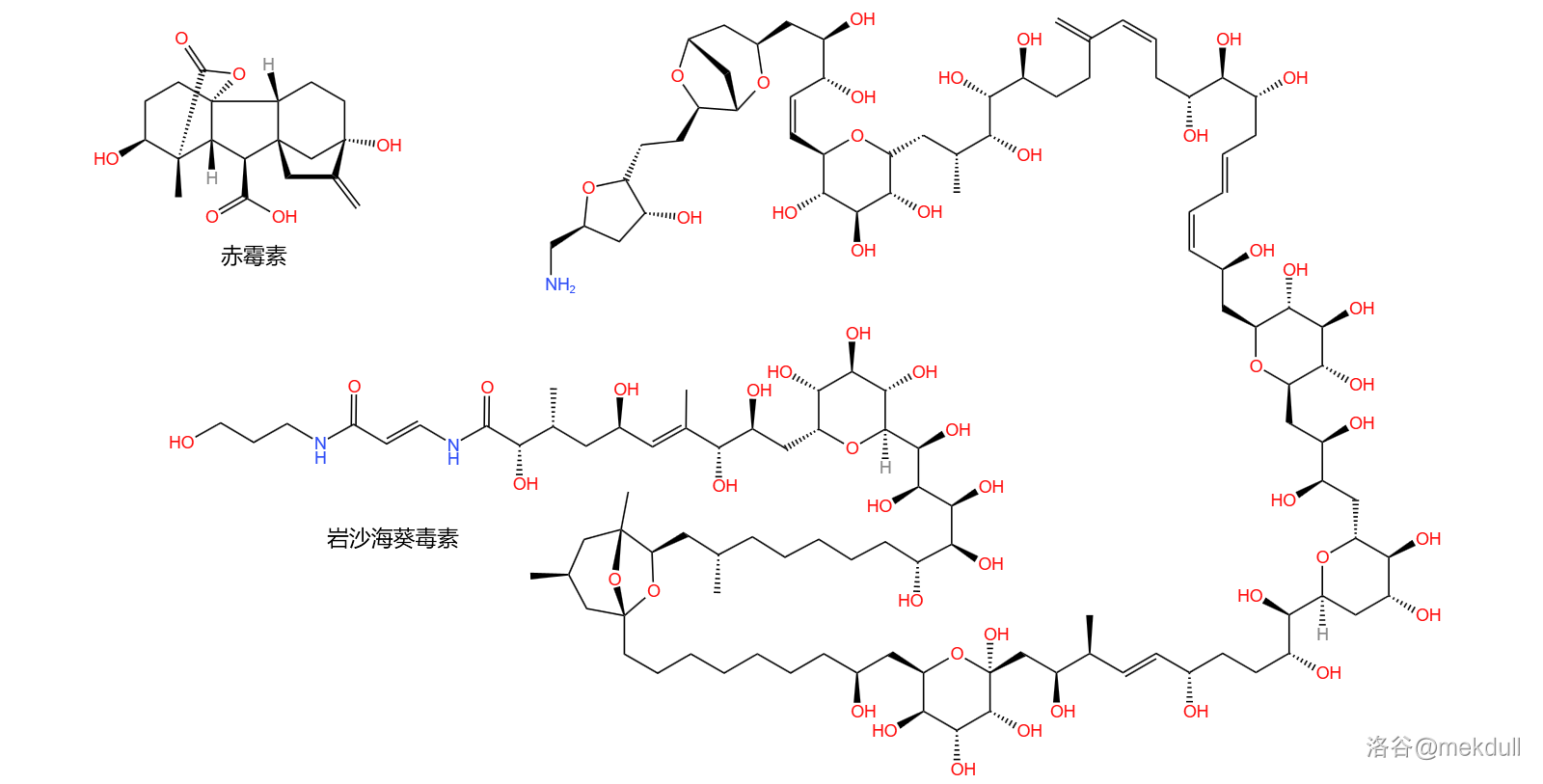
(超链接:关于岩沙海葵毒素全合成的详细解析)
相比于伍德沃德,另一位主角 $Eschenmoser$ 教授并没有在这次伟大的工作中收获多少名声,如今或许也没有多少人记得他的团队也是维生素B12全合成的重要参与者。更鲜有人知的是,在完成这项工作之后,他选择以维生素B12为切入点,研究两个根本看不到终点和希望的课题:生命的化学起源和演化对于天然产物的构建。尽管这注定是一条无比艰难的路,但他仍然义无反顾地走了下去。
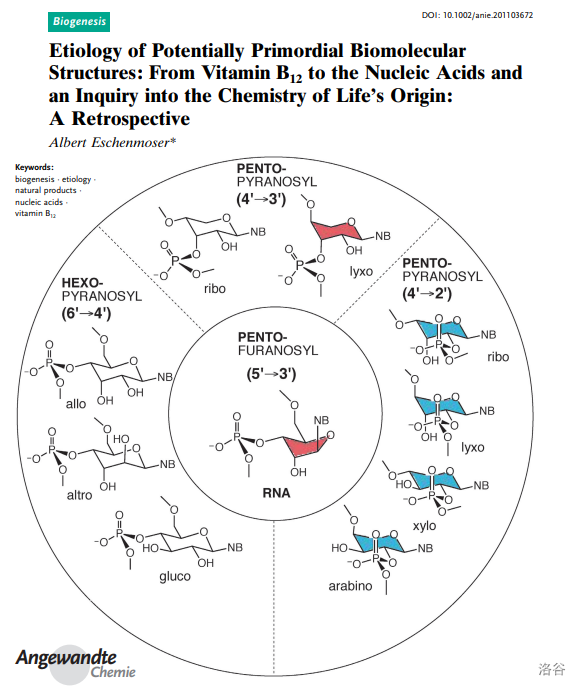
(上图:《潜在原始生物分子结构的病原学:从维生素B12到核酸,关于生命化学起源的探讨》论文影,$A.~Eschenmoser$,$2011$)
“我们可能永远也无从知晓”,这句老生常谈的话只会引导我们放弃寻找生命起源问题的努力······但对大自然创造生命之原理的探索永远不会真正结束,因为这是我们在探索我们自己的起源,探索我们自己的曾经。——《潜在原始生物分子结构的病原学:从维生素B12到核酸,关于生命化学起源的探讨》,$A.~Eschenmoser$,$2011$
$A.~Eschenmoser$ 于 $2023$ 年去世,享年 $98$ 岁。显然,直到去世他也没有找到这两个终极问题的答案。但是,他的学生们接替了他,在这条看不到尽头的道路上继续前行。
伍德沃德曾经说过一句流传很广的话:合成,是一门艺术。但我认为,不光是有机合成,任何科学探索,都是伟大的艺术。
而艺术,需要人们不断地去传承与创新。
5.参考文献
$[1]$:Woodward R B. The total synthesis of vitamin B12[J]. Pure and Applied Chemistry Eighth, 1973, 33(1): 145-178.
$[2]$:Eschenmoser A, Wintner C E. Natural Product Synthesis and Vitamin B12: Total synthesis of vitamin B12 provided a framework for exploration in several areas of organic chemistry[J]. Science, 1977, 196(4297): 1410-1420.
$[3]$:Woodward R B. Recent advances in the chemistry of natural products[J]. Pure and Applied Chemistry, 1968, 17(3-4): 519-547.
$[4]$:Eschenmoser A. Studies on organic synthesis[C]//XXIIIrd International Congress of Pure and Applied Chemistry: special lectures presented at Boston, USA, 26-30 July 1971. Butterworths, 1971, 2: 69-106.
$[5]$:Friedrich W, Gross G, Bernhauer K, et al. Synthesen auf dem Vitamin‐B12‐Gebiet. 4. Mitteilung Partialsynthese von Vitamin B12[J]. Helvetica Chimica Acta, 1960, 43(3): 704-712.
$[6]$:Eschenmoser A. Vitamin B12: experiments concerning the origin of its molecular structure[J]. Angewandte Chemie International Edition in English, 1988, 27(1): 5-39.
$[7]$:Eschenmoser A. Etiology of potentially primordial biomolecular structures: from vitamin B12 to the nucleic acids and an inquiry into the chemistry of life’s origin: a retrospective[J]. Angewandte Chemie International Edition, 2011, 50(52): 12412-12472.
$[8]$:Friedrich W, Bernhauer K. Zur Konstitution des Vitamin B12-Faktors III[J]. Zeitschrift für Naturforschung B, 1954, 9(11): 685-697.
$[9]$:Woodward R B, Hoffmann R. Stereochemistry of electrocyclic reactions [J]. J Am Chem Soc, 1965, 87(2): 395-397.
$[10]$:Woodward R B, Hoffmann R. Conservation of orbital symmetry [J]. Acc Chem Res, 1968, 1(1): 17-22.
$[11]$:Zimmerman H E. A mechanistic analysis of the Birch reduction[J]. Accounts of chemical research, 2012, 45(2): 164-170.
$[12]$:KARUVALAM R, HARIDAS K, SHETTY S. Trimethylsilyl chloride catalyzed synthesis of substituted benzimidazoles using two phase system under microwave conditions, and their antimicrobial studies[J]. Journal of the Chilean Chemical Society, 2012, 57(2): 1122-1125.
$[13]$:Blanche F, Cameron B, Crouzet J, et al. Vitamin B12: How the problem of its biosynthesis was solved [J]. Angew Chem Int Ed, 1995, 34(4): 383-411.